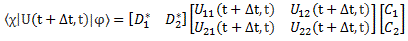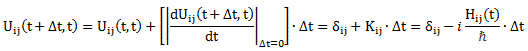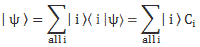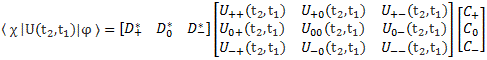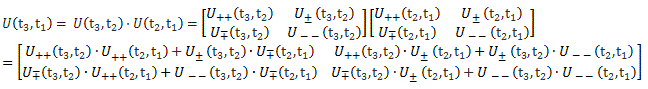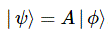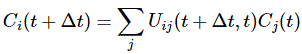Ammonia, i.e. NH3, is a colorless gas with a strong smell. Its serves as a precursor in the production of fertilizer, but we also know it as a cleaning product, ammonium hydroxide, which is NH3 dissolved in water. It has a lot of other uses too. For example, its use in this post, is to illustrate a two-state system. 🙂 We’ll apply everything we learned in our previous posts and, as I mentioned when finishing the last of those rather mathematical pieces, I think the example really feels like a reward after all of the tough work on all of those abstract concepts – like that Hamiltonian matrix indeed – so I hope you enjoy it. So… Here we go!
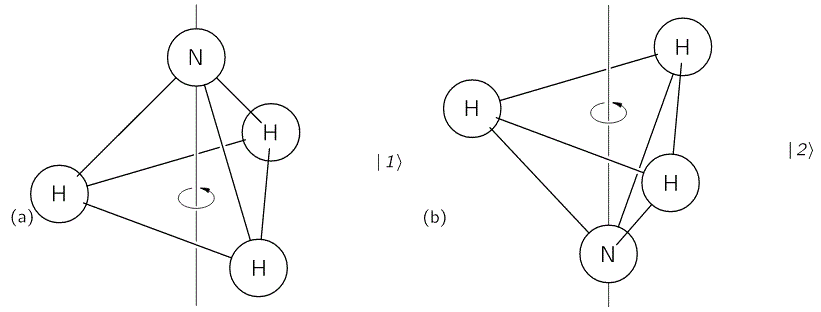
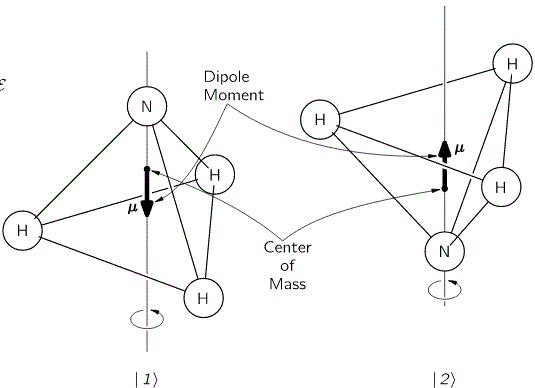
The geometry of the NH3 molecule can be described by thinking of it as a trigonal pyramid, with the nitrogen atom (N) at its apex, and the three hydrogen atoms (H) at the base, as illustrated below. [Feynman’s illustration is slightly misleading, though, because it may give the impression that the hydrogen atoms are bonded together somehow. That’s not the case: the hydrogen atoms share their electron with the nitrogen, thereby completing the outer shell of both atoms. This is referred to as a covalent bond. You may want to look it up, but it is of no particular relevance to what follows here.]
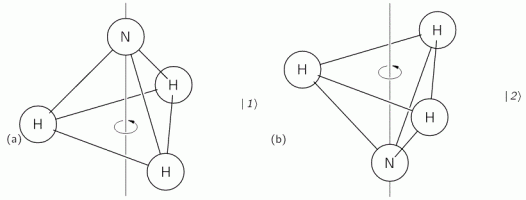
Here, we will only worry about the spin of the molecule about its axis of symmetry, as shown above, which is either in one direction or in the other, obviously. So we’ll discuss the molecule as a two-state system. So we don’t care about its translational (i.e. linear) momentum, its internal vibrations, or whatever else that might be going on. It is one of those situations illustrating that the spin vector, i.e. the vector representing angular momentum, is an axial vector: the first state, which is denoted by | 1 〉 is not the mirror image of state | 2 〉. In fact, there is a more sophisticated version of the illustration above, which usefully reminds us of the physics involved.
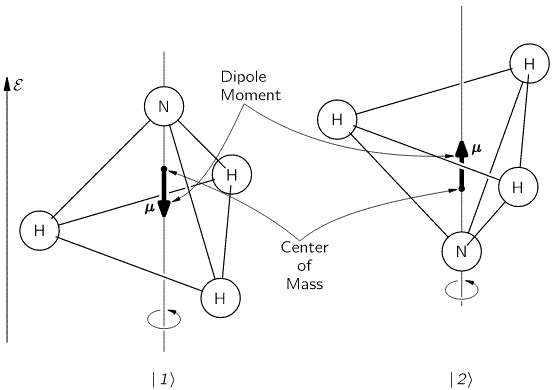
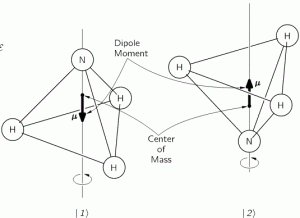 It should be noted, however, that we don’t need to specify what the energy barrier really consists of: moving the center of mass obviously requires some energy, but it is likely that a ‘flip’ also involves overcoming some electrostatic forces, as shown by the reversal of the electric dipole moment in the illustration above. In fact, the illustration may confuse you, because we’re usually thinking about some net electric charge that’s spinning, and so the angular momentum results in a magnetic dipole moment, that’s either ‘up’ or ‘down’, and it’s usually also denoted by the very same μ symbol that’s used below. As I explained in my post on angular momentum and the magnetic moment, it’s related to the angular momentum J through the so-called g-number. In the illustration above, however, the μ symbol is used to denote an electric dipole moment, so that’s different. Don’t rack your brain over it: just accept there’s an energy barrier, and it requires energy to get through it. Don’t worry about its details!
It should be noted, however, that we don’t need to specify what the energy barrier really consists of: moving the center of mass obviously requires some energy, but it is likely that a ‘flip’ also involves overcoming some electrostatic forces, as shown by the reversal of the electric dipole moment in the illustration above. In fact, the illustration may confuse you, because we’re usually thinking about some net electric charge that’s spinning, and so the angular momentum results in a magnetic dipole moment, that’s either ‘up’ or ‘down’, and it’s usually also denoted by the very same μ symbol that’s used below. As I explained in my post on angular momentum and the magnetic moment, it’s related to the angular momentum J through the so-called g-number. In the illustration above, however, the μ symbol is used to denote an electric dipole moment, so that’s different. Don’t rack your brain over it: just accept there’s an energy barrier, and it requires energy to get through it. Don’t worry about its details!
Indeed, in quantum mechanics, we abstract away from such nitty-gritty, and so we just say that we have base states | i 〉 here, with i equal to 1 or 2. One or the other. Now, in our post on quantum math, we introduced what Feynman only half-jokingly refers to as the Great Law of Quantum Physics: | = ∑ | i 〉〈 i | over all base states i. It basically means that we should always describe our initial and end states in terms of base states. Applying that principle to the state of our ammonia molecule, which we’ll denote by | ψ 〉, we can write:
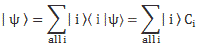
You may – in fact, you should – mechanically apply that | = ∑ | i 〉〈 i | substitution to | ψ 〉 to get what you get here, but you should also think about what you’re writing. It’s not an easy thing to interpret, but it may help you to think of the similarity of the formula above with the description of a vector in terms of its base vectors, which we write as A = Ax·e1 + Ay·e2 + Az·e3. Just substitute the Ai coefficients for Ci and the ei base vectors for the | i 〉 base states, and you may understand this formula somewhat better. It also explains why the | ψ 〉 state is often referred to as the | ψ 〉 state vector: unlike our A = ∑ Ai·ei sum of base vectors, our | 1 〉 C1 + | 2 〉 C2 sum does not have any geometrical interpretation but… Well… Not all ‘vectors’ in math have a geometric interpretation, and so this is a case in point.
It may also help you to think of the time-dependency. Indeed, this formula makes a lot more sense when realizing that the state of our ammonia molecule, and those coefficients Ci, depend on time, so we write: ψ = ψ(t) and Ci = Ci(t). Hence, if we would know, for sure, that our molecule is always in state | 1 〉, then C1 = 1 and C2 = 0, and we’d write: | ψ 〉 = | 1 〉 = | 1 〉 1 + | 2 〉 0. [I am always tempted to insert a little dot (·), and change the order of the factors, so as to show we’re talking some kind of product indeed – so I am tempted to write | ψ 〉 = C1·| 1 〉 C1 + C2·| 2 〉 C2, but I note that’s not done conventionally, so I won’t do it either.]
Why this time dependency? It’s because we’ll allow for the possibility of the nitrogen to push its way through the pyramid – through the three hydrogens, really – and flip to the other side. It’s unlikely, because it requires a lot of energy to get half-way through (we’ve got what we referred to as an energy barrier here), but it may happen and, as we’ll see shortly, it results in us having to think of the the ammonia molecule as having two separate energy levels, rather than just one. We’ll denote those energy levels as E0 ± A. However, I am getting ahead of myself here, so let me get back to the main story.
To fully understand the story, you should really read my previous post on the Hamiltonian, which explains how those Ci coefficients, as a function of time, can be determined. They’re determined by a set of differential equations (i.e. equations involving a function and the derivative of that function) which we wrote as:

If we have two base states only – which is the case here – then this set of equations is:
Two equations and two functions – C1 = C1(t) and C2 = C2(t) – so we should be able to solve this thing, right? Well… No. We don’t know those Hij coefficients. As I explained in my previous post, they also evolve in time, so we should write them as Hij(t) instead of Hij tout court, and so it messes the whole thing up. We have two equations and six functions really. There is no way we can solve this! So how do we get out of this mess?
Well… By trial and error, I guess. 🙂 Let us just assume the molecule would behave nicely—which we know it doesn’t, but so let’s push the ‘classical’ analysis as far as we can, so we might get some clues as to how to solve this problem. In fact, our analysis isn’t ‘classical’ at all, because we’re still talking amplitudes here! However, you’ll agree the ‘simple’ solution would be that our ammonia molecule doesn’t ‘tunnel’. It just stays in the same spin direction forever. Then H12 and H21 must be zero (think of the U12(t + Δt, t) and U21(t + Δt, t) functions) and H11 and H22 are equal to… Well… I’d love to say they’re equal to 1 but… Well… You should go through my previous posts: these Hamiltonian coefficients are related to probabilities but… Well… Same-same but different, as they say in Asia. 🙂 They’re amplitudes, which are things you use to calculate probabilities. But calculating probabilities involve normalization and other stuff, like allowing for interference of amplitudes, and so… Well… To make a long story short, if our ammonia molecule would stay in the same spin direction forever, then H11 and H22 are not one but some constant. In any case, the point is that they would not change in time (so H11(t) = H11 and H22(t ) = H22), and, therefore, our two equations would reduce to:

So the coefficients are now proper coefficients, in the sense that they’ve got some definite value, and so we have two equations and two functions only now, and so we can solve this. Indeed, remembering all of the stuff we wrote on the magic of exponential functions (more in particular, remembering that d[ex]/dx), we can understand the proposed solution:
As Feynman notes: “These are just the amplitudes for stationary states with the energies E1 = H11 and E2 = H22.” Now let’s think about that. Indeed, I find the term ‘stationary’ state quite confusing, as it’s ill-defined. In this context, it basically means that we have a wavefunction that is determined by (i) a definite (i.e. unambiguous, or precise) energy level and (ii) that there is no spatial variation. Let me refer you to my post on the basics of quantum math here. We often use a sort of ‘Platonic’ example of the wavefunction indeed:
a·e−i·θ = e−i·(ω·t − k ∙x) = a·e−(i/ħ)·(E·t − p∙x)
So that’s a wavefunction assuming the particle we’re looking at has some well-defined energy E and some equally well-defined momentum p. Now, that’s kind of ‘Platonic’ indeed, because it’s more like an idea, rather than something real. Indeed, a wavefunction like that means that the particle is everywhere and nowhere, really—because its wavefunction is spread out all of over space. Of course, we may think of the ‘space’ as some kind of confined space, like a box, and then we can think of this particle as being ‘somewhere’ in that box, and then we look at the temporal variation of this function only – which is what we’re doing now: we don’t consider the space variable x at all. So then the equation reduces to a·e–(i/ħ)·(E·t), and so… Well… Yes. We do find that our Hamiltonian coefficient Hii is like the energy of the | i 〉 state of our NH3 molecule, so we write: H11 = E1, and H22 = E2, and the ‘wavefunctions’ of our C1 and C2 coefficients can be written as:
- C1 = a·e−(i/ħ)·(H11·t) = a·e−(i/ħ)·(E1·t), with H11 = E1, and
- C2 = a·e−(i/ħ)·(H22·t) = a·e−(i/ħ)·(E2·t), with H22 = E2.
But can we interpret C1 and C2 as proper amplitudes? They are just coefficients in these equations, aren’t they? Well… Yes and no. From what we wrote in previous posts, you should remember that these Ci coefficients are equal to 〈 i | ψ 〉, so they are the amplitude to find our ammonia molecule in one state or the other.
Back to Feynman now. He adds, logically but brilliantly:
“We note, however, that for the ammonia molecule the two states |1〉 and |2〉 have a definite symmetry. If nature is at all reasonable, the matrix elements H11 and H22 must be equal. We’ll call them both E0, because they correspond to the energy the states would have if H11 and H22 were zero.”
So our C1 and C2 amplitudes then reduce to:
- C1 = 〈 1 | ψ 〉 = a·e−(i/ħ)·(E0·t)
- C2 =〈 2 | ψ 〉 = a·e−(i/ħ)·(E0·t)
We can now take the absolute square of both to find the probability for the molecule to be in state 1 or in state 2:
- |〈 1 | ψ 〉|2 = |a·e−(i/ħ)·(E0·t)|2 = a2
- |〈 2 | ψ 〉|2 = |a·e−(i/ħ)·(E0·t)|2 = a2
Now, the probabilities have to add up to 1, so a2 + a2 = 1 and, therefore, the probability to be in either in state 1 or state 2 is 0.5, which is what we’d expect.
Note: At this point, it is probably good to get back to our | ψ 〉 = | 1 〉 C1 + | 2 〉 C2 equation, so as to try to understand what it really says. Substituting the a·e−(i/ħ)·(E0·t) expression for C1 and C2 yields:
| ψ 〉 = | 1 〉 a·e−(i/ħ)·(E0·t) + | 2 〉 a·e−(i/ħ)·(E0·t) = [| 1 〉 + | 2 〉] a·e−(i/ħ)·(E0·t)
Now, what is this saying, really? In our previous post, we explained this is an ‘open’ equation, so it actually doesn’t mean all that much: we need to ‘close’ or ‘complete’ it by adding a ‘bra’, i.e. a state like 〈 χ |, so we get a 〈 χ | ψ〉 type of amplitude that we can actually do something with. Now, in this case, our final 〈 χ | state is either 〈 1 | or 〈 2 |, so we write:
- 〈 1 | ψ 〉 = [〈 1 | 1 〉 + 〈 1 | 2 〉]·a·e−(i/ħ)·(E0·t) = [1 + 0]·a·e−(i/ħ)·(E0·t)· = a·e−(i/ħ)·(E0·t)
- 〈 2 | ψ 〉 = [〈 2 | 1 〉 + 〈 2 | 2 〉]·a·e−(i/ħ)·(E0·t) = [0 + 1]·a·e−(i/ħ)·(E0·t)· = a·e−(i/ħ)·(E0·t)
Note that I finally added the multiplication dot (·) because we’re talking proper amplitudes now and, therefore, we’ve got a proper product too: we multiply one complex number with another. We can now take the absolute square of both to find the probability for the molecule to be in state 1 or in state 2:
- |〈 1 | ψ 〉|2 = |a·e−(i/ħ)·(E0·t)|2 = a2
- |〈 2 | ψ 〉|2 = |a·e−(i/ħ)·(E0·t)|2 = a2
Unsurprisingly, we find the same thing: these probabilities have to add up to 1, so a2 + a2 = 1 and, therefore, the probability to be in state 1 or state 2 is 0.5. So the notation and the logic behind makes perfect sense. But let me get back to the lesson now.
The point is: the true meaning of a ‘stationary’ state here, is that we have non-fluctuating probabilities. So they are and remain equal to some constant, i.e. 1/2 in this case. This implies that the state of the molecule does not change: there is no way to go from state 1 to state 2 and vice versa. Indeed, if we know the molecule is in state 1, it will stay in that state. [Think about what normalization of probabilities means when we’re looking at one state only.]
You should note that these non-varying probabilities are related to the fact that the amplitudes have a non-varying magnitude. The phase of these amplitudes varies in time, of course, but their magnitude is and remains a, always. The amplitude is not being ‘enveloped’ by another curve, so to speak.
OK. That should be clear enough. Sorry I spent so much time on this, but this stuff on ‘stationary’ states comes back again and again and so I just wanted to clear that up as much as I can. Let’s get back to the story.
So we know that, what we’re describing above, is not what ammonia does really. As Feynman puts it: “The equations [i.e. the C1 and C2 equations above] don’t tell us what what ammonia really does. It turns out that it is possible for the nitrogen to push its way through the three hydrogens and flip to the other side. It is quite difficult; to get half-way through requires a lot of energy. How can it get through if it hasn’t got enough energy? There is some amplitude that it will penetrate the energy barrier. It is possible in quantum mechanics to sneak quickly across a region which is illegal energetically. There is, therefore, some [small] amplitude that a molecule which starts in |1〉 will get to the state |2〉. The coefficients H12 and H21 are not really zero.”
He adds: “Again, by symmetry, they should both be the same—at least in magnitude. In fact, we already know that, in general, Hij must be equal to the complex conjugate of Hji.”
His next step, then, is to interpreted as either a stroke of genius or, else, as unexplained. 🙂 He invokes the symmetry of the situation to boldly state that H12 is some real negative number, which he denotes as −A, which – because it’s a real number (so the imaginary part is zero) – must be equal to its complex conjugate H21. So then Feynman does this fantastic jump in logic. First, he keeps using the E0 value for H11 and H22, motivating that as follows: “If nature is at all reasonable, the matrix elements H11 and H22 must be equal, and we’ll call them both E0, because they correspond to the energy the states would have if H11 and H22 were zero.” Second, he uses that minus A value for H12 and H21. In short, the two equations and six functions are now reduced to:

Solving these equations is rather boring. Feynman does it as follows:
Now, what does these equations actually mean? It depends on those a and b coefficients. Looking at the solutions, the most obvious question to ask is: what if a or b are zero? If b is zero, then the second terms in both equations is zero, and so C1 and C2 are exactly the same: two amplitudes with the same temporal frequency ω = (E0 − A)/ħ. If a is zero, then C1 and C2 are the same too, but with opposite sign: two amplitudes with the same temporal frequency ω = (E0 + A)/ħ. Squaring them – in both cases (i.e. for a = 0 or b = 0) – yields, once again, an equal and constant probability for the spin of the ammonia molecule to in the ‘up’ or ‘down’ or ‘down’. To be precise, we We can now take the absolute square of both to find the probability for the molecule to be in state 1 or in state 2:
- For b = 0: |〈 1 | ψ 〉|2 = |(a/2)·e−(i/ħ)·(E0 − A)·t|2 = a2/4 = |〈 2 | ψ 〉|2
- For a = 0: |〈 1 | ψ 〉|2 =|(b/2)·e−(i/ħ)·(E0 + A)·t|2 = b2/4 = |〈 2 | ψ 〉|2 (the minus sign in front of b/2 is squared away)
So we get two stationary states now. Why two instead of one? Well… You need to use your imagination a bit here. They actually reflect each other: they’re the same as the one stationary state we found when assuming our nitrogen atom could not ‘flip’ from one position to the other. It’s just that the introduction of that possibility now results in a sort of ‘doublet’ of energy levels. But so we shouldn’t waste our time on this, as we want to analyze the general case, for which the probabilities to be in state 1 or state 2 do vary in time. So that’s when a and b are non-zero.
To analyze it all, we may want to start with equating t to zero. We then get:

This leads us to conclude that a = b = 1, so our equations for C1(t) and C2(t) can now be written as:
Remembering our rules for adding and subtracting complex conjugates (eiθ + e–iθ = 2cosθ and eiθ − e–iθ = 2sinθ), we can re-write this as:
Now these amplitudes are much more interesting. Their temporal variation is defined by E0 but, on top of that, we have an envelope here: the cos(A·t/ħ) and sin(A·t/ħ) factor respectively. So their magnitude is no longer time-independent: both the phase as well as the amplitude now vary with time. What’s going on here becomes quite obvious when calculating and plotting the associated probabilities, which are
- |C1(t)|2 = cos2(A·t/ħ), and
- |C2(t)|2 = sin2(A·t/ħ)
respectively (note that the absolute square of i is equal to 1, not −1). The graph of these functions is depicted below.

As Feynman puts it: “The probability sloshes back and forth.” Indeed, the way to think about this is that, if our ammonia molecule is in state 1, then it will not stay in that state. In fact, one can be sure the nitrogen atom is going to flip at some point in time, with the probabilities being defined by that fluctuating probability density function above. Indeed, as time goes by, the probability to be in state 2 increases, until it will effectively be in state 2. And then the cycle reverses.
Our | ψ 〉 = | 1 〉 C1 + | 2 〉 C2 equation is a lot more interesting now, as we do have a proper mix of pure states now: we never really know in what state our molecule will be, as we have these ‘oscillating’ probabilities now, which we should interpret carefully.
The point to note is that the a = 0 and b = 0 solutions came with precise temporal frequencies: (E0 − A)/ħ and (E0 + A)/ħ respectively, which correspond to two separate energy levels: E0 − A and E0 + A respectively, with |A| = H12 = H21. So everything is related to everything once again: allowing the nitrogen atom to push its way through the three hydrogens, so as to flip to the other side, thereby breaking the energy barrier, is equivalent to associating two energy levels to the ammonia molecule as a whole, thereby introducing some uncertainty, or indefiniteness as to its energy, and that, in turn, gives us the amplitudes and probabilities that we’ve just calculated.
Note that the probabilities “sloshing back and forth”, or “dumping into each other” – as Feynman puts it – is the result of the varying magnitudes of our amplitudes, going up and down and, therefore, their absolute square varies too.
So… Well… That’s it as an introduction to a two-state system. There’s more to come. Ammonia is used in the ammonia maser. Now that is something that’s interesting to analyze—both from a classical as well as from a quantum-mechanical perspective. Feynman devotes a full chapter to it, so I’d say… Well… Have a look. 🙂
Post scriptum: I must assume this analysis of the NH3 molecule, with the nitrogen ‘flipping’ across the hydrogens, triggers a lot of questions, so let me try to answer some. Let me first insert the illustration once more, so you don’t have to scroll up:
The first thing that you should note is that the ‘flip’ involves a change in the center of mass position. So that requires energy, which is why we associate two different energy levels with the molecule: E0 + A and E0 − A. However, as mentioned above, we don’t care about the nitty-gritty here: the energy barrier is likely to combine a number of factors, including electrostatic forces, as evidenced by the flip in the electric dipole moment, which is what the μ symbol here represents! Just note that the two energy levels are separated by an amount that’s equal to 2·A, rather than A and that, once again, it becomes obvious now why Feynman would prefer the Hamiltonian to be called the ‘energy matrix’, as its coefficients do represent specific energy levels, or differences between them! Now, that assumption yielded the following wavefunctions for C1 = 〈 1 | ψ 〉 and C1 = 〈 2 | ψ 〉:
- C1 = 〈 1 | ψ 〉 = (1/2)·e−(i/ħ)·(E0 − A)·t + (1/2)·e−(i/ħ)·(E0 + A)·t
- C2 = 〈 2 | ψ 〉 = (1/2)·e−(i/ħ)·(E0 − A)·t – (1/2)·e−(i/ħ)·(E0 + A)·t
Both are composite waves. To be precise, they are the sum of two component waves with a temporal frequency equal to ω1 = (E0 − A)/ħ and ω1 = (E0 + A)/ħ respectively. [As for the minus sign in front of the second term in the wave equation for C2, −1 = e±iπ, so + (1/2)·e−(i/ħ)·(E0 + A)·t and – (1/2)·e−(i/ħ)·(E0 + A)·t are the same wavefunction: they only differ because their relative phase is shifted by ±π.]
Now, writing things this way, rather than in terms of probabilities, makes it clear that the two base states of the molecule themselves are associated with two different energy levels, so it is not like one state has more energy than the other. It’s just that the possibility of going from one state to the other requires an uncertainty about the energy, which is reflected by the energy doublet E0 ± A in the wavefunction of the base states. Now, if the wavefunction of the base states incorporates that energy doublet, then it is obvious that the state of the ammonia molecule, at any point in time, will also incorporate that energy doublet.
This triggers the following remark: what’s the uncertainty really? Is it an uncertainty in the energy, or is it an uncertainty in the wavefunction? I mean: we have a function relating the energy to a frequency. Introducing some uncertainty about the energy is mathematically equivalent to introducing uncertainty about the frequency. Think of it: two energy levels implies two frequencies, and vice versa. More in general, introducing n energy levels, or some continuous range of energy levels ΔE, amounts to saying that our wave function doesn’t have a specific frequency: it now has n frequencies, or a range of frequencies Δω = ΔE/ħ. Of course, the answer is: the uncertainty is in both, so it’s in the frequency and in the energy and both are related through the wavefunction. So… In a way, we’re chasing our own tail.
Having said that, the energy may be uncertain, but it is real. It’s there, as evidenced by the fact that the ammonia molecule behaves like an atomic oscillator: we can excite it in exactly the same way as we can excite an electron inside an atom, i.e. by shining light on it. The only difference is the photon energies: to cause a transition in an atom, we use photons in the optical or ultraviolet range, and they give us the same radiation back. To cause a transition in an ammonia molecule, we only need photons with energies in the microwave range. Here, I should quickly remind you of the frequencies and energies involved. visible light is radiation in the 400–800 terahertz range and, using the E = h·f equation, we can calculate the associated energies of a photon as 1.6 to 3.2 eV. Microwave radiation – as produced in your microwave oven – is typically in the range of 1 to 2.5 gigahertz, and the associated photon energy is 4 to 10 millionths of an eV. Having illustrated the difference in terms of the energies involved, I should add that masers and lasers are based on the same physical principle: LASER and MASER stand for Light/Micro-wave Amplification by Stimulated Emission of Radiation, respectively.
So… How shall I phrase this? There’s uncertainty, but the way we are modeling that uncertainty matters. So yes, the uncertainty in the frequency of our wavefunction and the uncertainty in the energy are mathematically equivalent, but the wavefunction has a meaning that goes much beyond that. [You may want to reflect on that yourself.]
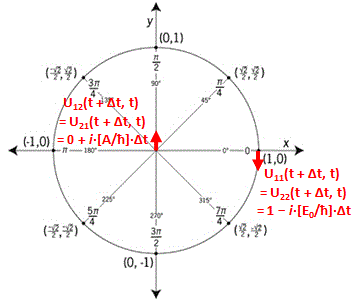
Finally, another question you may have is why would Feynman take minus A (i.e. −A) for H12 and H21. Frankly, my first thought on this was that it should have something to do with the original equation for these Hamiltonian coefficients, which also has a minus sign: Uij(t + Δt, t) = δij + Kij(t)·Δt = δij − (i/ħ)·Hij(t)·Δt. For i ≠ j, this reduces to:
Uij(t + Δt, t) = + Kij(t)·Δt = − (i/ħ)·Hij(t)·Δt
However, the answer is: it really doesn’t matter. One could write: H12 and H21 = +A, and we’d find the same equations. We’d just switch the indices 1 and 2, and the coefficients a and b. But we get the same solutions. You can figure that out yourself. Have fun with it !
Oh ! And please do let me know if some of the stuff above would trigger other questions. I am not sure if I’ll be able to answer them, but I’ll surely try, and good question always help to ensure we sort of ‘get’ this stuff in a more intuitive way. Indeed, when everything is said and done, the goal of this blog is not simply re-produce stuff, but to truly ‘get’ it, as good as we can. 🙂
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 17, 2020 as a result of a DMCA takedown notice from Michael A. Gottlieb, Rudolf Pfeiffer, and The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 17, 2020 as a result of a DMCA takedown notice from Michael A. Gottlieb, Rudolf Pfeiffer, and The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 20, 2020 as a result of a DMCA takedown notice from Michael A. Gottlieb, Rudolf Pfeiffer, and The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 20, 2020 as a result of a DMCA takedown notice from Michael A. Gottlieb, Rudolf Pfeiffer, and The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/