
Pre-script (dated 26 June 2020): This post has become less relevant (even irrelevant, perhaps) because my views on all things quantum-mechanical have evolved significantly as a result of my progression towards a more complete realist (classical) interpretation of quantum physics. The text also got mutilated because of the removal of material by the dark force. I keep blog posts like these mainly because I want to keep track of where I came from. I might review them one day, but I currently don’t have the time or energy for it. 🙂
Original post:
Phew! I am quite happy I got through Feynman’s chapters on thermodynamics. Now is a good time to review the math behind it. We thoroughly understand the gas equation now:
PV = NkT = (γ–1)U
The gamma (γ) in this equation is the specific heat ratio: it’s 5/3 for ideal gases (so that’s about 1.667) and, theoretically, 4/3 ≈ 1.333 or 9/7 ≈ 1.286 for diatomic gases, depending on the degrees of freedom we associate with diatomic molecules. More complicated molecules have even more degrees of freedom and, hence, can absorb even more energy, so γ gets closer to one—according to the kinetic gas theory, that is. While we know that the kinetic gas theory is not quite accurate – an approach involving molecular energy states is a better match for reality – that doesn’t matter here. As for the term (specific heat ratio), I’ll explain that later. [I promise. 🙂 You’ll see it’s quite logical.]
The point to note is that this body of gas (or whatever substance) stores an amount of energy U that is directly proportional to the temperature (T), and Nk/(γ–1) is the constant of proportionality. We can also phrase it the other way around: the temperature is directly proportional to the energy, with (γ–1)/Nk the constant of proportionality. It means temperature and energy are in a linear relationship. [Yes, direct proportionality implies linearity.] The graph below shows the T = [(γ–1)/Nk]·U relationship for three different values of γ, ranging from 5/3 (i.e. the maximum value, which characterizes monatomic noble gases such as helium, neon or krypton) to a value close to 1, which is characteristic of more complicated molecular arrangements indeed, such as heptane (γ = 1.06) or methyl butane ((γ = 1.08). The illustration shows that, unlike monatomic gas, more complicated molecular arrangements allow the gas to absorb a lot of (heat) energy with a relatively moderate rise in temperature only.
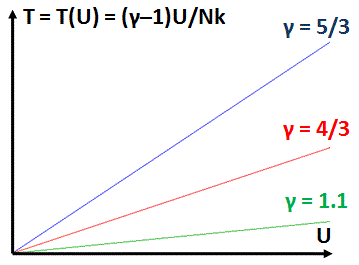 We’ll soon encounter another variable, enthalpy (H), which is also linearly related to energy: H = γU. From a math point of view, these linear relationships don’t mean all that much: they just show these variables – temperature, energy and enthalphy – are all directly related and, hence, can be defined in terms of each other.
We’ll soon encounter another variable, enthalpy (H), which is also linearly related to energy: H = γU. From a math point of view, these linear relationships don’t mean all that much: they just show these variables – temperature, energy and enthalphy – are all directly related and, hence, can be defined in terms of each other.
We can invent other variables, like the Gibbs energy, or the Helmholtz energy. In contrast, entropy, while often being mentioned as just some other state function, is something different altogether. In fact, the term ‘state function’ causes a lot of confusion: pressure and volume are state variables too. The term is used to distinguish these variables from so-called process functions, notably heat and work. Process functions describe how we go from one equilibrium state to another, as opposed to the state variables, which describe the equilibrium situation itself. Don’t worry too much about the distinction—for now, that is.
Let’s look at non-linear stuff. The PV = NkT = (γ–1)U says that pressure (P) and volume (V) are inversely proportional one to another, and so that’s a non-linear relationship. [Yes, inverse proportionality is non-linear.] To help you visualize things, I inserted a simple volume-pressure diagram below, which shows how pressure and volume are related for three different values of U (or, what amounts to the same, three different values of T).
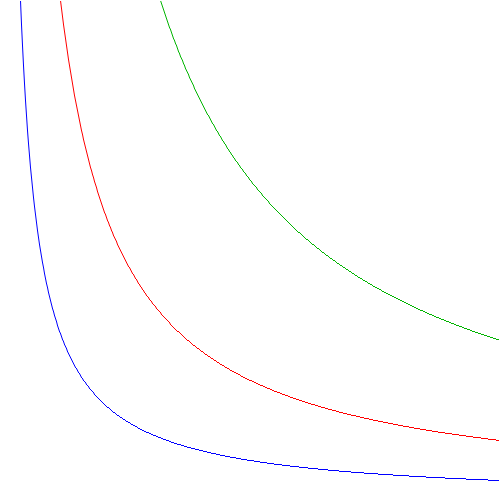
The curves are simple hyperbolas which have the x- and y-axis as horizontal and vertical asymptote respectively. If you’ve studied social sciences (like me!) – so if you know a tiny little bit of the ‘dismal science’, i.e. economics (like me!) – you’ll note they look like indifference curves. The x- and y-axis then represent the quantity of some good X and some good Y respectively, and the curves closer to the origin are associated with lower utility. How much X and Y we will buy then, depends on (a) their price and (b) our budget, which we represented by a linear budget line tangent to the curve we can reach with our budget, and then we are a little bit happy, very happy or extremely happy, depending on our budget. Hence, our budget determines our happiness. From a math point of view, however, we can also look at it the other way around: our happiness determines our budget. [Now that‘s a nice one, isn’t it? Think about it! 🙂 And, in the process, think about hyperbolas too: the y = 1/x function holds the key to understanding both infinity and nothingness. :-)]
U is a state function but, as mentioned above, we’ve got quite a few state variables in physics. Entropy, of course, denoted by S—and enthalpy too, denoted by H. Let me remind you of the basics of the entropy concept:
- The internal energy U changes because (a) we add or remove some heat from the system (ΔQ), (b) because some work is being done (by the gas on its surroundings or the other way around), or (c) because of both. Using the differential notation, we write: dU = dQ – dW, always. The (differential) work that’s being done is PdV. Hence, we have dU = dQ – PdV.
- When transferring heat to a system at a certain temperature, there’s a quantity we refer to as the entropy. Remember that illustration of Feynman’s in my post on entropy: we go from one point to another on the temperature-volume diagram, taking infinitesimally small steps along the curve, and, at each step, an infinitesimal amount of work dW is done, and an infinitesimal amount of entropy dS = dQ/T is being delivered.
- The total change in entropy, ΔS, is a line integral: ΔS = ∫L dQ/T = ∫L dS.
That’s somewhat tougher to understand than economics, and so that’s why it took me more time to come with terms with it. 🙂 Just go through Feynman’s Lecture on it, or through that post I referenced above. If you don’t want to do that, then just note that, while entropy is a very mysterious concept, it’s deceptively simple from a math point of view: ΔS = ΔQ/T, so the (infinitesimal) change in entropy is, quite simply, the ratio of (1) the (infinitesimal or incremental) amount of heat that is being added or removed as the system goes from one state to another through a reversible process and (2) the temperature at which the heat is being transferred. However, I am not writing this post to discuss entropy once again. I am writing it to give you an idea of the math behind the system.
So dS = dQ/T. Hence, we can re-write dU = dQ – dW as:
dU = TdS – PdV ⇔ dU + d(PV) = TdS – PdV + d(PV)
⇔ d(U + PV) = dH = TdS – PdV + PdV + VdP = TdS + VdP
The U + PV quantity on the left-hand side of the equation is the so-called enthalpy of the system, which I mentioned above. It’s denoted by H indeed, and it’s just another state variable, like energy: same-same but different, as they say in Asia. We encountered it in our previous post also, where we said that chemists prefer to analyze the behavior of substances using temperature and pressure as ‘independent variables’, rather than temperature and volume. Independent variables? What does that mean, exactly?
According to the PV = NkT equation, we only have two independent variables: if we assign some value to two variables, we’ve got a value for the third one. Indeed, remember that other equation we got when we took the total differential of U. We wrote U as U(V, T) and, taking the total differential, we got:
dU = (∂U/∂T)dT + (∂U/∂V)dV
We did not need to add a (∂U/∂P)dP term, because the pressure is determined by the volume and the temperature. We could also have written U = U(P, T) and, therefore, that dU = (∂U/∂T)dT + (∂U/∂P)dP. However, when working with temperature and pressure as the ‘independent’ variables, it’s easier to work with H rather than U. The point to note is that it’s all quite flexible really: we have two independent variables in the system only. The third one (and all of the other variables really, like energy or enthalpy or whatever) depend on the other two. In other words, from a math point of view, we only have two degrees of freedom in the system here: only two variables are actually free to vary. 🙂
Let’s look at that dH = TdS + VdP equation. That’s a differential equation in which not temperature and pressure, but entropy (S) and pressure (P) are ‘independent’ variables, so we write:
dH(S, P) = TdS + VdP
Now, it is not very likely that we will have some problem to solve with data on entropy and pressure. At our level of understanding, any problem that’s likely to come our way will probably come with data on more common variables, such as the heat, the pressure, the temperature, and/or the volume. So we could continue with the expression above but we don’t do that. It makes more sense to re-write the expression substituting TdS for dQ once again, so we get:
dH = dQ + VdP
That resembles our dU = dQ – PdV expression: it just substitutes V for –P. And, yes, you guessed it: it’s because the two expressions resemble each other that we like to work with H now. 🙂 Indeed, we’re talking the same system and the same infinitesimal changes and, therefore, we can use all the formulas we derived already by just substituting H for U, V for –P, and dP for dV. Huh? Yes. It’s a rather tricky substitution. If we switch V for –P (or vice versa) in a partial derivative involving T, we also need to include the minus sign. However, we do not need to include the minus sign when substituting dV and dP, and we also don’t need to change the sign of the partial derivatives of U and H when going from one expression to another! It’s a subtle and somewhat weird point, but a very important one! I’ll explain it in a moment. Just continue to read as for now. Let’s do the substitution using our rules:
dU = (∂Q/∂T)VdT + [T(∂P/∂T)V − P]dV becomes:
dH = (∂Q/∂T)PdT + (∂H/∂P)TdP = CPdT + [–T·(∂V/∂T)P + V]dP
Note that, just as we referred to (∂Q/∂T)V as the specific heat capacity of a substance at constant volume, which we denoted by CV, we now refer to (∂Q/∂T)P as the specific heat capacity at constant pressure, which we’ll denote, logically, as CP. Dropping the subscripts of the partial derivatives, we re-write the expression above as:
dH = CPdT + [–T·(∂V/∂T) + V]dP
So we’ve got what we wanted: we switched from an expression involving derivatives assuming constant volume to an expression involving derivatives assuming constant pressure. [In case you wondered what we wanted, this is it: we wanted an equation that helps us to solve another type of problem—another formula for a problem involving a different set of data.]
As mentioned above, it’s good to use subscripts with the partial derivatives to emphasize what changes and what is constant when calculating those partial derivatives but, strictly speaking, it’s not necessary, and you will usually not find the subscripts when googling other texts. For example, in the Wikipedia article on enthalpy, you’ll find the expression written as:
dH = CPdT + V(1–αT)dP with α = (1/V)(∂V/∂T)
Just write it all out and you’ll find it’s the same thing, exactly. It just introduces another coefficient, α, i.e. the coefficient of (cubic) thermal expansion. If you find this formula is easier to remember, then please use this one. It doesn’t matter.
Now, let’s explain that funny business with the minus signs in the substitution. I’ll do so by going back to that infinitesimal analysis of the reversible cycle in my previous post, in which we had that formula involving ΔQ for the work done by the gas during an infinitesimally small reversible cycle: ΔW = ΔVΔP = ΔQ·(ΔT/T). Now, we can either write that as:
- ΔQ = T·(ΔP/ΔT)·ΔV = dQ = T·(∂P/∂T)V·dV – which is what we did for our analysis of (∂U/∂V)T – or, alternatively, as
- ΔQ = T·(ΔV/ΔT)·ΔP = dQ = T·(∂V/∂T)P·dP, which is what we’ve got to do here, for our analysis of (∂H/∂P)T.
Hence, dH = dQ + VdP becomes dH = T·(∂V/∂T)P·dP + V·dP, and dividing all by dP gives us what we want to get: dH/dP = (∂H/∂P)T = T·(∂V/∂T)P + V.
[…] Well… NO! We don’t have the minus sign in front of T·(∂V/∂T)P, so we must have done something wrong or, else, that formula above is wrong.
The formula is right (it’s in Wikipedia, so it must be right :-)), so we are wrong. Indeed! The thing is: substituting dT, dV and dP for ΔT, ΔV and ΔP is somewhat tricky. The geometric analysis (illustrated below) makes sense but we need to watch the signs.
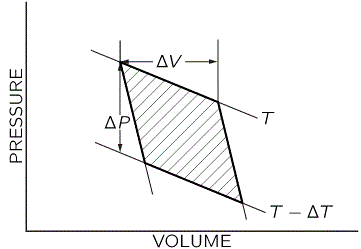
We’ve got a volume increase, a temperature drop and, hence, also a pressure drop over the cycle: the volume goes from V to V+ΔV (and then back to V, of course), while the pressure and the temperature go from P to P–ΔP and T to T–ΔT respectively (and then back to P and T, of course). Hence, we should write: ΔV = dV, –ΔT = dT, and –ΔP = dP. Therefore, as we replace the ratio of the infinitesimal change of pressure and temperature, ΔP/ΔT, by a proper derivative (i.e. ∂P/∂T), we should add a minus sign: ΔP/ΔT = –∂P/∂T. Now that gives us what we want: dH/dP = (∂H/∂P)T = –T·(∂V/∂T)P + V, and, therefore, we can, indeed, write what we wrote above:
dU = (∂Q/∂T)VdT + [T(∂P/∂T)V − P]dV becomes:
dH = (∂Q/∂T)PdT + [–T·(∂V/∂T)P + V]dP = CPdT + [–T·(∂V/∂T)P + V]dP
Now, in case you still wonder: what’s the use of all these different expressions stating the same? The answer is simple: it depends on the problem and what information we have. Indeed, note that all derivatives we use in our expression for dH expression assume constant pressure, so if we’ve got that kind of data, we’ll use the chemists’ representation of the system. If we’ve got data describing performance at constant volume, we’ll need the physicists’ formulas, which are given in terms of derivatives assuming constant volume. It all looks complicated but, in the end, it’s the same thing: the PV = NkT equation gives us two ‘independent’ variables and one ‘dependent’ variable. Which one is which will determine our approach.
Now, we left one thing unexplained. Why do we refer to γ as the specific heat ratio? The answer is: it is the ratio of the specific heat capacities indeed, so we can write:
γ = CP/CV
However, it is important to note that that’s valid for ideal gases only. In that case, we know that the (∂U/∂V)P derivative in our dU = (∂U/∂T)VdT + (∂U/∂V)TdV expression is zero: we can change the volume, but if the temperature remains the same, the internal energy remains the same. Hence, dU = (∂U/∂T)VdT = CVdT, and dU/dT = CV. Likewise, the (∂H/∂P)T derivative in our dH = (∂H/∂T)PdT + (∂H/∂P)TdP expression is zero—for ideal gases, that is. Hence, dH = (∂H/∂T)PdT = CPdT, and dH/dT = CP. Hence,
CP/CV = (dH/dT)/(dU/dT) = dH/dU
Does that make sense? If dH/dU = γ, then H must be some linear function of U. More specifically, H must be some function H = γU + c, with c some constant (it’s the so-called constant of integration). Now, γ is supposed to be constant too, of course. That’s all perfectly fine: indeed, combining the definition of H (H = U + PV), and using the PV = (γ–1)U relation, we have H = U + (γ–1)U = γU (hence, c = 0). So, yes, dH/dU = γ, and γ = CP/CV.
Note the qualifier, however: we’re assuming γ is constant (which does not imply the gas has to be ideal, so the interpretation is less restrictive than you might think it is). If γ is not a constant, it’s a different ballgame. […] So… Is γ actually constant? The illustration below shows γ is not constant for common diatomic gases like hydrogen or (somewhat less common) oxygen. It’s the same for other gases: when mentioning γ, we need to state the temperate at which we measured it too. 😦 However, the illustration also shows the assumption of γ being constant holds fairly well if temperature varies only slightly (like plus or minus 100° C), so that’s OK. 🙂

I told you so: the kinetic gas theory is not quite accurate. An approach involving molecular energy states works much better (and is actually correct, as it’s consistent with quantum theory). But so we are where we are and I’ll save the quantum-theoretical approach for later. 🙂
So… What’s left? Well… If you’d google the Wikipedia article on enthalphy in order to check if I am not writing nonsense, you’ll find it gives γ as the ratio of H and U itself: γ = H/U. That’s not wrong, obviously (γ = H/U = γU/U = γ), but that formula doesn’t really explain why γ is referred to as the specific heat ratio, which is what I wanted to do here.
OK. We’ve covered a lot of ground, but let’s reflect some more. We did not say a lot about entropy, and/or the relation between energy and entropy. Too bad… The relationship between entropy and energy is obviously not so simple as between enthalpy and energy. Indeed, because of that easy H = γU relationship, enthalpy emerges as just some auxiliary variable: some temporary variable we need to calculate something. Entropy is, obviously, something different. Unlike enthalpy, entropy involves very complicated thinking, involving (ir)reversibility and all that. So it’s quite deep, I’d say – but I’ll write more about that later. I think this post has gone as far as it should. 🙂
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
