
Pre-script (dated 26 June 2020): This post has become less relevant (even irrelevant, perhaps) because my views on all things quantum-mechanical have evolved significantly as a result of my progression towards a more complete realist (classical) interpretation of quantum physics. The text also got mutilated because of the removal of material by the dark force. I keep blog posts like these mainly because I want to keep track of where I came from. I might review them one day, but I currently don’t have the time or energy for it. 🙂
Original post:
In my previous posts, it was mentioned repeatedly that the kinetic theory of gases is not quite correct: the experimentally measured values of the so-called specific heat ratio (γ) vary with temperature and, more importantly, their values differ, in general, from what classical theory would predict. It works, more or less, for noble gases, which do behave as ideal gases and for which γ is what the kinetic theory of gases would want it to be: γ = 5/3—but we get in trouble immediately, even for simple diatomic gases like oxygen or hydrogen, as illustrated below: the theoretical value is 9/7 (so that’s 1.286, more or less), but the measured value is very different.
 Let me quickly remind you how we get the theoretical number. According to classical theory, a diatomic molecule like oxygen can be represented as two atoms connected by a spring. Each of the atoms absorbs kinetic energy, and for each direction of motion (x, y and z), that energy is equal to kT/2, so the kinetic energy of both atoms – added together – is 2·3·kT/2 = 3kT. However, I should immediately add that not all of that energy is to be associated with the center-of-mass motion of the whole molecule, which determines the temperature of the gas: that energy is and remains equal to the 3kT/2, always. We also have rotational and vibratory motion. The molecule can rotate in two independent directions (and any combination of these directions, of course) and, hence, rotational motion is to absorb an amount of energy equal to 2·kT/2 = kT. Finally, the vibratory motion is to be analyzed as any other oscillation, so like a spring really. There is only one dimension involved and, hence, the kinetic energy here is just kT/2. However, we know that the total energy in an oscillator is the sum of the kinetic and potential energy, which adds another kT/2 term. Putting it all together, we find that the average energy for each diatomic particle is (or should be) equal to 7·kT/2 = (7/2)kT. Now, as mentioned above, the temperature of the gas (T) is proportional to the mean molecular energy of the center-of-mass motion only (in fact, that’s how temperature is defined), with the constant of proportionality equal to 3k/2. Hence, for monatomic ideal gases, we can write: U = N·(3k/2)T and, therefore, PV = NkT = (2/3)·U. Now, γ appears as follows in the ideal gas law: PV = (γ–1)U. Therefore, γ = 2/3 + 1 = 5/3, but so that’s for monatomic ideal gases only! The total kinetic energy of our diatomic molecule is U = N·(7k/2)T and, therefore, PV = (2/7)·U. So γ must be γ = 2/7 + 1 = 9/7 ≈ 1.286 for diatomic gases, like oxygen and hydrogen.
Let me quickly remind you how we get the theoretical number. According to classical theory, a diatomic molecule like oxygen can be represented as two atoms connected by a spring. Each of the atoms absorbs kinetic energy, and for each direction of motion (x, y and z), that energy is equal to kT/2, so the kinetic energy of both atoms – added together – is 2·3·kT/2 = 3kT. However, I should immediately add that not all of that energy is to be associated with the center-of-mass motion of the whole molecule, which determines the temperature of the gas: that energy is and remains equal to the 3kT/2, always. We also have rotational and vibratory motion. The molecule can rotate in two independent directions (and any combination of these directions, of course) and, hence, rotational motion is to absorb an amount of energy equal to 2·kT/2 = kT. Finally, the vibratory motion is to be analyzed as any other oscillation, so like a spring really. There is only one dimension involved and, hence, the kinetic energy here is just kT/2. However, we know that the total energy in an oscillator is the sum of the kinetic and potential energy, which adds another kT/2 term. Putting it all together, we find that the average energy for each diatomic particle is (or should be) equal to 7·kT/2 = (7/2)kT. Now, as mentioned above, the temperature of the gas (T) is proportional to the mean molecular energy of the center-of-mass motion only (in fact, that’s how temperature is defined), with the constant of proportionality equal to 3k/2. Hence, for monatomic ideal gases, we can write: U = N·(3k/2)T and, therefore, PV = NkT = (2/3)·U. Now, γ appears as follows in the ideal gas law: PV = (γ–1)U. Therefore, γ = 2/3 + 1 = 5/3, but so that’s for monatomic ideal gases only! The total kinetic energy of our diatomic molecule is U = N·(7k/2)T and, therefore, PV = (2/7)·U. So γ must be γ = 2/7 + 1 = 9/7 ≈ 1.286 for diatomic gases, like oxygen and hydrogen.
Phew! So that’s the theory. However, as we can see from the diagram, γ approaches that value only when we heat the gas to a few thousand degrees! So what’s wrong? One assumption is that certain kinds of motions “freeze out” as the temperature falls—although it’s kinda weird to think of something ‘freezing out’ at a thousand degrees Kelvin! In any case, at the end of the 19th century, that was the assumption that was advanced, very reluctantly, by scientists such as James Jeans. However, the mystery was about to be solved then, as Max Planck, even more reluctantly, presented his quantum theory of energy at the turn of the century itself.
But the quantum theory was confirmed and so we should now see how we can apply it to the behavior of gas. In my humble view, it’s a really interesting analysis, because we’re applying quantum theory here to a phenomenon that’s usually being analyzed as a classical problem only.
Boltzmann’s Law
We derived Boltzmann’s Law in our post on the First Principles of Statistical Mechanics. To be precise, we gave Boltzmann’s Law for the density of a gas (which we denoted by n = N/V) in a force field, like a gravitational field, or in an electromagnetic field (assuming our gas particles are electrically charged, of course). We noted, however, Boltzmann’s Law was also applicable to much more complicated situations, like the one below, which shows a potential energy function for two molecules that is quite characteristic of the way molecules actually behave: when they come very close together, they repel each other but, at larger distances, there’s a force of attraction. We don’t really know the forces behind but we don’t need to: as long as these forces are conservative, they can combine in whatever way they want to combine, and Boltzmann’s Law will be applicable. [It should be obvious why. If you hesitate, just think of the definition of work and how it affects potential energy and all that. Work is force times distance, but when doing work, we’re also changing potential energy indeed! So if we’ve got a potential energy function, we can get all the rest.]
Boltzmann’s Law itself is illustrated by the graph below, which also gives the formula for it: n = n0·e−P.E/kT.
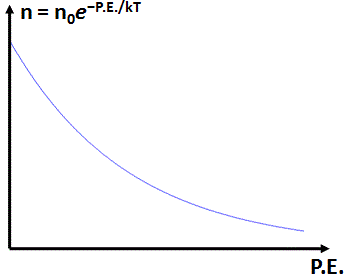
It’s a graph starting at n = n0 for P.E. = 0, and it then decreases exponentially. [Funny expression, isn’t it? So as to respect mathematical terminology, I should say that it decays exponentially.] In any case, if anything, Boltzmann’s Law shows the natural exponential function is quite ‘natural’ indeed, because Boltzmann’s Law pops up in Nature everywhere! Indeed, Boltzmann’s Law is not limited to functions of potential energy only. For example, Feynman derives another Boltzmann Law for the distribution of molecular speeds or, so as to ensure the formula is also valid in relativity, the distribution of molecular momenta. In case you forgot, momentum (p) is the product of mass (m) and velocity (u), and the relevant Boltzmann Law is:
f(p)·dp = C·e−K.E/kT·dp
The argument is not terribly complicated but somewhat lengthy, and so I’ll refer you to the link for more details. As for the f(p) function (and the dp factor on both sides of the equation), that’s because we’re not talking exact values of p but some range equal to dp and some probability of finding particles that have a momentum within that range. The principle is illustrated below for molecular speeds (denoted by u = p/m), so we have a velocity distribution below. The illustration for p would look the same: just substitute u for p.
Boltzmann’s Law can be stated, much more generally, as follows:
The probability of different conditions of energy (E), potential or kinetic, is proportional to e−E/kT.
As Feynman notes, “This is a rather beautiful proposition, and a very easy thing to remember too!” It is, and we’ll need it for the next bit.
The quantum-mechanical theory of gases
According to quantum theory, energy comes in discrete packets, quanta, and any system, like an oscillator, will only have a discrete set of energy levels, i.e. states of different energy. An energy state is, obviously, a condition of energy and, hence, Boltzmann’s Law applies. More specifically, if we denote the various energy levels, i.e. the energies of the various molecular states, by E0, E1, E2,…, Ei,…, and if Boltzmann’s Law applies, then the probability of finding a molecule in the particular state Ei will be proportional to e−Ei /kT.
Now, we know we’ve got some constant there, but we can get rid of that by calculating relative probabilities. For example, the probability of being in state E1, relative to the probability of being in state E0, is:
P1/P0 = e−E1 /kT/e−E0 /kT = e−(E1–E0)/kT
But the relative probability P1 should, obviously, also be equal to the ratio n1/N, i.e. the ratio of the number of molecules in state E1 and the total number of molecules. Likewise, P0 = n0/N. Hence, P1/P0 = n1/n0 and, therefore, we can write:
n = n0e−(E1–E0)/kT
What can we do with that? Remember we want to explain the behavior of non-monatomic gas—like diatomic gas, for example. Now we need some other assumption, obviously. As it turns out, the assumption that we can represent a system as some kind of oscillation still makes sense! In fact, the assumption that our diatomic molecule is like a spring is equally crucial to our quantum-theoretical analysis of gases as it is to our classical kinetic theory of gases. To be precise, in both theories, we look at it as a harmonic oscillator.
Don’t panic. A harmonic oscillator is, quite simply, a system that, when displaced from its equilibrium position, experiences some kind of restoring force. Now, for it to be harmonic, the force needs to be linear. For example, when talking springs, the restoring force F will be proportional to the displacement x). It basically means we can use a linear differential equation to analyze the system, like m·(d2x/dt2) = –kx. […] I hope you recognize this equation, because you should! It’s Newton’s Law: F = m·a with F = –k·x. If you remember the equation, you’ll also remember that harmonic oscillations were sinusoidal oscillations with a constant amplitude and a constant frequency. That frequency did not depend on the amplitude: because of the sinusoidal function involved, it was easier to write that frequency as an angular frequency, which we denoted by ω0 and which, in the case of our spring, was equal to ω0 = (k/m)1/2. So it’s a property of the system. Indeed, ω0 is the square root of the ratio of (1) k, which characterizes the spring (it’s its stiffness), and (2) m, i.e. the mass on the spring. Solving the differential equation yielded x = A·cos(ω0t + Δ) as a general solution, with A the (maximum) amplitude, and Δ some phase shift determined by our t = 0 point. Let me quickly jot down too more formulas: the potential energy in the spring is kx2/2, while its kinetic energy is mv2/2, as usual (so the kinetic energy depends on the mass and its velocity, while the potential energy only depends on the displacement and the spring’s stiffness). Of course, kinetic and potential energy add up to the total energy of the system, which is constant and proportional to the square of the (maximum) amplitude: K.E. + P.E. = E ∝ A2. To be precise, E = kA2/2.
That’s simple enough. Let’s get back to our molecular oscillator. While the total energy of an oscillator in classical theory can take on any value, Planck challenged that assumption: according to quantum theory, it can only take up energies equal to ħω at a time. [Note that we use the so-called reduced Planck constant here (i.e. h-bar), because we’re dealing with angular frequencies.] Hence, according to quantum theory, we have an oscillator with equally spaced energy levels, and the difference between them is ħω. Now, ħω is terribly tiny—but it’s there. Let me visualize what I just wrote:
So our expression for P1/P0 becomes P1/P0 = e−ħω/kT/e−0/kT = e−ħω/kT. More generally, we have Pi/P0 = e−i·ħω/kT. So what? Well… We’ve got a function here which gives the chance of finding a molecule in state Pi relative to that of finding it in state E0, and it’s a function of temperature. Now, the graph below illustrates the general shape of that function. It’s a bit peculiar, but you can see that the relative probability goes up and down with temperature. The graph makes it clear that, at extremely low temperatures, most particles will be in state E0 and, of course, the internal energy of our body of gas will be close to nil.
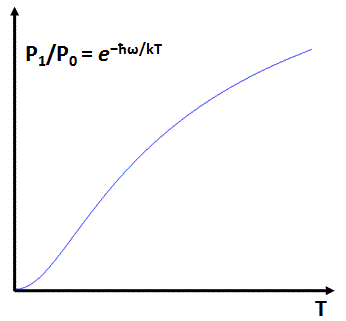
Now, we can look at the oscillators in the bottom state (i.e. particles in the molecular energy state E0) as being effectively ‘frozen’: they don’t contribute to the specific heat. However, as we increase the temperature, our molecules gradually begin to have an appreciable probability to be in the second state, and then in the next state, and so on, and so the internal energy of the gas increases effectively. Now, when the probability is appreciable for many states, the quantized states become nearly indistinguishable and, hence, the situation is like classical physics: it is nearly indistinguishable from a continuum of energies.
Now, while you can imagine such analysis should explain why the specific heat ratio for oxygen and hydrogen varies as it does in the very first graph of this post, you can also imagine the details of that analysis fill quite a few pages! In fact, even Feynman doesn’t include it in his Lectures. What he does include is the analysis of the blackbody radiation problem, which is remarkably similar. So… Well… For more details on that, I’ll refer you to Feynman indeed. 🙂
I hope you appreciated this little ‘lecture’, as it sort of wraps up my ‘series’ of posts on statistical mechanics, thermodynamics and, central to both, the classical theory of gases. Have fun with it all!
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:

I’m trying to understand this page after a relevant Twitter convo today, and I have to take it slowly if I want any hope of understanding. To start, I’m already stuck on the second paragraph :P:
>According to classical theory, a diatomic molecule like oxygen can be represented as two atoms >connected by a spring. Each of the atoms absorbs kinetic energy, and for each direction of >motion (x, y and z), that energy is equal to kT/2, so the kinetic energy of both atoms – added >together – is 2·3·kT/2 = 3kT.
Where did you get kT/2, and why are you able to sum x, y, z directions of kinetic energy as if they were components of a vector (Let vector v = v_x + v_y. Since kinetic energy isn’t a vector, k_v_x + k_v_y != k_v)?
> The molecule can rotate in two independent directions (and any combination of these directions, of course) and, hence, rotational motion is to absorb an amount of energy equal to 2·kT/2 = kT.
Same question here- are you able to sum rotational energies separately and get the correct total rotational energy?
Where do you get two directions from? Even if the two molecules are attached by a spring, I can visualize creating > 2 arbitrary axes of rotation and the molecule _as a whole_ (i.e. not the individual atoms) rotating about all these axes at once.
>Putting it all together, we find that the average energy for each diatomic particle is (or should be) >equal to 7·kT/2 = (7/2)kT.
Where did 7/2 come from? I saw 2·3·kT/2 (kinetic) + 2 * kT/2 (rotational) + kT/2 (oscillator- kinetic) + kT/2 (oscillator- potential) = 10*kT/2. How did I end up with 3 extra “kT/2” terms?
Hi William – I’ve done a few posts on this. The basic one (from June 2015, see: https://readingfeynman.org/2015/06/24/first-principles-of-statistical-mechanics/) is followed by a number of more sophisticated ones (including the one you are stuck on), so you may want to check all of the ‘archives’ of June and July 2015, so the two or three ones that precede the one you’re stuck at. It is all very interesting because – as you point out – energy is supposed to be a non-directional quantity (it’s a scalar quantity, not a vector quantity). However, the analysis involves the calculation of the average kinetic energy of molecules and, therefore, the average velocity (so that is 〈m·v2/2〉 instead of mv2/2) – and velocity is a vector quantity with an x, y and z direction. The 1/3 factor is there because the average velocity in the x-, y- and z-direction are all the same and uncorrelated, so 〈vx2〉 = 〈vy2〉 = 〈vz2〉 = [〈vx2〉 + 〈vy2〉 + 〈vz2〉]/3 = 〈v2〉/3. [Sorry I can’t do subscripts and superscripts here.] In any case… Have a look. The various coefficients (2/3 or 7/2) have to do with the degrees of freedom. For mono-atomic gases, molecules can only vibrate in the x, y and z directions. But diatomic molecules have, effectively, additional degrees of freedom. The post I refer to above has the analysis: we distinguish between the velocities of atom A and atom B, and from there… Well… Have a look and see if you have any questions left. Be patient with yourself because I remember a good understanding of those degrees of freedom didn’t come easily to me either ! When both rotational and linear movement is involved, it becomes kinda weird to see what’s truly ‘independent’ motion. But you’ll get there ! [I got there and I am just your average Joe… So… Well… It just takes time and perseverance.] Cheers and have fun ! – Jean Louis