Pre-scriptum added much later: We have advanced much in our understanding since we wrote this post. If you are reading it because you want to understand more about the boson-fermion distinction, then you shouldn’t be here. The general distinction between bosons and fermions is a useless theoretical generalization which actually prevents you from understanding what is really going on. I am keeping this post online for documentation purposes only. It is interesting from a math point of view but you are not here to learn math, are you?
Jean Louis Van Belle, 20 May 2020
Original post:
I’ve discussed statistics, in the context of quantum mechanics, a couple of times already (see, for example, my post on amplitudes and statistics). However, I never took the time to properly explain those distribution functions which are referred to as the Maxwell-Boltzmann, Bose-Einstein and Fermi-Dirac distribution functions respectively. Let me try to do that now—without, hopefully, getting lost in too much math! It should be a nice piece, as it connects quantum mechanics with statistical mechanics, i.e. two topics I had nicely separated so far. 🙂
You know the Boltzmann Law now, which says that the probabilities of different conditions of energy are given by e−energy/kT = 1/eenergy/kT. Different ‘conditions of energy’ can be anything: density, molecular speeds, momenta, whatever. The point is: we have some probability density function f, and it’s a function of the energy E, so we write:
f(E) = C·e−energy/kT = C/eenergy/kT
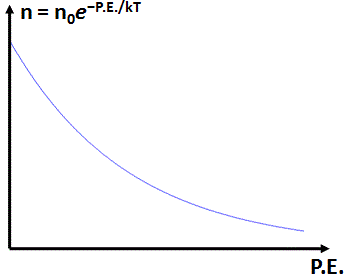
C is just a normalization constant (all probabilities have to add up to one, so the integral of this function over its domain must be one), and k and T are also usual suspects: T is the (absolute) temperature, and k is the Boltzmann constant, which relates the temperate to the kinetic energy of the particles involved. We also know the shape of this function. For example, when we applied it to the density of the atmosphere at various heights (which are related to the potential energy, as P.E. = m·g·h), assuming constant temperature, we got the following graph. The shape of this graph is that of an exponential decay function (we’ll encounter it again, so just take a mental note of it).

A more interesting application is the quantum-mechanical approach to the theory of gases, which I introduced in my previous post. To explain the behavior of gases under various conditions, we assumed that gas molecules are like oscillators but that they can only take on discrete levels of energy. [That’s what quantum theory is about!] We denoted the various energy levels, i.e. the energies of the various molecular states, by E0, E1, E2,…, Ei,…, and if Boltzmann’s Law applies, then the probability of finding a molecule in the particular state Ei is proportional to e−Ei /kT. We can then calculate the relative probabilities, i.e. the probability of being in state Ei, relative to the probability of being in state E0, is:
Pi/P0 = e−Ei /kT/e−E0 /kT = e−(Ei–E0)/kT = 1/e(Ei–E0)/kT
Now, Pi obviously equals ni/N, so it is the ratio of the number of molecules in state Ei (ni) and the total number of molecules (N). Likewise, P0 = n0/N and, therefore, we can write:
ni/n0 = e−(Ei−E0)/kT = 1/e(Ei–E0)/kT
This formulation is just another Boltzmann Law, but it’s nice in that it introduces the idea of a ground state, i.e. the state with the lowest energy level. We may or may not want to equate E0 with zero. It doesn’t matter really: we can always shift all energies by some arbitrary constant because we get to choose the reference point for the potential energy.
So that’s the so-called Maxwell-Boltzmann distribution. Now, in my post on amplitudes and statistics, I had jotted down the formulas for the other distributions, i.e. the distributions when we’re not talking classical particles but fermions and/or bosons. As you know, fermions are particles governed by the Fermi exclusion principle: indistinguishable particles cannot be together in the same state. For bosons, it’s the other way around: having one in some quantum state actually increases the chance of finding another one there, and we can actually have an infinite number of them in the same state.
We also know that fermions and bosons are the real world: fermions are the matter-particles, bosons are the force-carriers, and our ‘Boltzmann particles’ are nothing but a classical approximation of the real world. Hence, even if we can’t see them in the actual world, the Fermi-Dirac and Bose-Einstein distributions are the real-world distributions. 🙂 Let me jot down the equations once again:
Fermi-Dirac (for fermions): f(E) = 1/[Ae(E − EF)/kT + 1]
Bose-Einstein (for bosons): f(E) = 1/[AeE/kT − 1]
We’ve got some other normalization constant here (A), which we shouldn’t be too worried about—for the time being, that is. Now, to see how these distributions are different from the Maxwell-Boltzmann distribution (which we should re-write as f(E) = C·e−E/kT = 1/[A·eE/kT] so as to make all formulas directly comparable), we should just make a graph. Please go online to find a graph tool (I found a new one recently—really easy to use), and just do it. You’ll see they are all like that exponential decay function. However, in order to make a proper comparison, we would actually need to calculate the normalization coefficients and, for the Fermi energy, we would also need the Fermi energy EF (note that, for simplicity, we did equate E0 with zero). Now, we could give it a try, but it’s much easier to google and find an example online.
The HyperPhysics website of Georgia State University gives us one: the example assumes 6 particles and 9 energy levels, and the table and graph below compare the Maxwell-Boltzmann and Bose-Einstein distributions for the model.

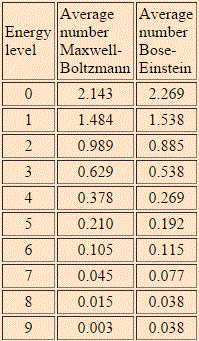
Now that is an interesting example, isn’t it? In this example (but all depends on its assumptions, of course), the Maxwell-Boltzmann and Bose-Einstein distributions are almost identical. Having said that, we can clearly see that the lower energy states are, indeed, more probable with Bose-Einstein statistics than with the Maxwell-Boltzmann statistics. While the difference is not dramatic at all in this example, the difference does become very dramatic, in reality, with large numbers (i.e. high matter density) and, more importantly, at very low temperatures, at which bosons can condense into the lowest energy state. This phenomenon is referred to as Bose-Einstein condensation: it causes superfluidity and superconductivity, and it’s real indeed: it has been observed with supercooled He-4, which is not an everyday substance, but real nevertheless!
What about the Fermi-Dirac distribution for this example? The Fermi-Dirac distribution is given below: the lowest energy state is now less probable, the mid-range energies much more, and none of the six particles occupy any of the four highest energy levels. Again, while the difference is not dramatic in this example, it can become very dramatic, in reality, with large numbers (read: high matter density) and very low temperatures: at absolute zero, all of the possible energy states up to the Fermi energy level will be occupied, and all the levels above the Fermi energy will be vacant.
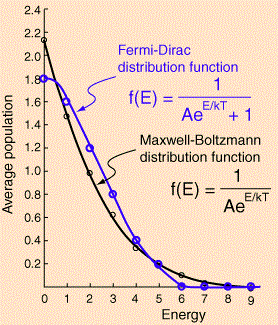
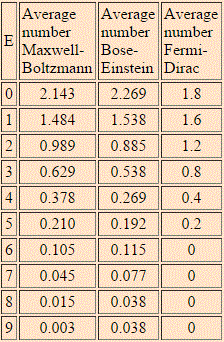
What can we make out of all of this? First, you may wonder why we actually have more than one particle in one state above: doesn’t that contradict the Fermi exclusion principle? No. We need to distinguish micro- and macro-states. In fact, the example assumes we’re talking electrons here, and so we can have two particles in each energy state—with opposite spin, however. At the same time, it’s true we cannot have three, or more, in any state. That results, in the example we’re looking at here, in five possible distributions only, as shown below.
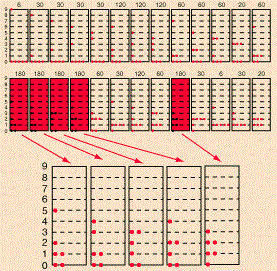
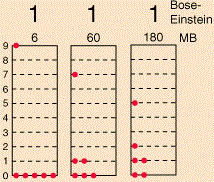
The diagram is an interesting one: if the particles were to be classical particles, or bosons, then 26 combinations are possible, including the five Fermi-Dirac combinations, as shown above. Note the little numbers above the 26 possible combinations (e.g. 6, 20, 30,… 180): they are proportional to the likelihood of occurring under the Maxwell-Boltzmann assumption (so if we assume the particles are ‘classical’ particles). Let me introduce you to the math behind the example by using the diagram below, which shows three possible distributions/combinations (I know the terminology is quite confusing—sorry for that!).
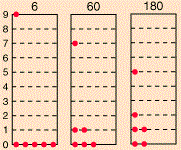
If we could distinguish the particles, then we’d have 2002 micro-states, which is the total of all those little numbers on top of the combinations that are shown (6+60+180+…). However, the assumption is that we cannot distinguish the particles. Therefore, the first combination in the diagram above, with five particles in the zero energy state and one particle in state 9, occurs 6 times into 2002 and, hence, it has a probability of 6/2002 ≈ 0.003 only. In contrast, the second combination is 10 times more likely, and the third one is 30 times more likely! In any case, the point is, in the classical situation (and in the Bose-Einstein hypothesis as well), we have 26 possible macro-states, as opposed to 5 only for fermions, and so that leads to a very different density function. Capito?
No? Well, this blog is not a textbook on physics and, therefore, I should refer you to the mentioned site once again, which references a 1992 textbook on physics (Frank Blatt, Modern Physics, 1992) as the source of this example. However, I won’t do that: you’ll find the details in the Post Scriptum to this post. 🙂
Let’s first focus on the fundamental stuff, however. The most burning question is: if the real world consists of fermions and bosons, why is that that we only see the Maxwell-Boltzmann distribution in our actual (non-real?) world? 🙂 The answer is that both the Fermi-Dirac and Bose-Einstein distribution approach the Maxwell–Boltzmann distribution if higher temperatures and lower particle densities are involved. In other words, we cannot see the Fermi-Dirac distributions (all matter is fermionic, except for weird stuff like superfluid helium-4 at 1 or 2 degrees Kelvin), but they are there!
Let’s approach it mathematically: the most general formula, encompassing both Fermi-Dirac and Bose-Einstein statistics, is:
Ni(Ei) ∝ 1/[e(Ei − μ)/kT ± 1]
If you’d google, you’d find a formula involving an additional coefficient, gi, which is the so-called degeneracy of the energy level Ei. I included it in the formula I used in the above-mentioned post of mine. However, I don’t want to make it any more complicated than it already is and, therefore, I omitted it this time. What you need to look at are the two terms in the denominator: e(Ei − μ)/kT and ± 1.
From a math point of view, it is obvious that the values of e(Ei − μ)/kT + 1 (Fermi-Dirac) and e(Ei − μ)/kT − 1 (Bose-Einstein) will approach each other if e(Ei − μ)/kT is much larger than ±1, so if e(Ei − μ)/kT >> 1. That’s the case, obviously, if the (Ei − μ)/kT ratio is large, so if (Ei − μ) >> kT. In fact, (Ei − μ) should, obviously, be much larger than kT for the lowest energy levels too! Now, the conditions under which that is the case are associated with the classical situation (such as a cylinder filled with gas, for example). Why?
Well… […] Again, I have to say that this blog can’t substitute for a proper textbook. Hence, I am afraid I have to leave it to you to do the necessary research to see why. 🙂 The non-mathematical approach is to simple note that quantum effects, i.e. the ±1 term, only apply if the concentration of particles is high enough. Indeed, quantum effects appear if the concentration of particles is higher than the so-called quantum concentration. Only when the quantum concentration is reached, particles will start interacting according to what they are, i.e. as bosons or as fermions. At higher temperature, that concentration will not be reached, except in massive objects such as a white dwarf (white dwarfs are stellar remnants with the mass like that of the Sun but a volume like that of the Earth). So, in general, we can say that at higher temperatures and at low concentration we will not have any quantum effects. That should settle the matter—as for now, at least.
You’ll have one last question: we derived Boltzmann’s Law from the kinetic theory of gases, but how do we derive that Ni(Ei) = 1/[Ae(Ei − μ)/kT ± 1] expression? Good question but, again, we’d need more than a few pages to explain that! The answer is: quantum mechanics, of course! Go check it out in Feynman’s third Volume of Lectures! 🙂
Post scriptum: combinations, permutations and multiplicity
The mentioned example from HyperPhysics is really interesting, if only because it shows you also need to master a bit of combinatorics to get into quantum mechanics. Let’s go through the basics. If we have n distinct objects, we can order hem in n! ways, with n! (read: n factorial) equal to n·(n–1)·(n–2)·…·3·2·1. Note that 0! is equal to 1, per definition. We’ll need that definition.
For example, a red, blue and green ball can be ordered in 3·2·1 = 6 ways. Each way is referred to as a permutation.
Besides permutations, we also have the concept of a k-permutation, which we can denote in a number of ways but let’s choose P(n, k). [The P stands for permutation here, not for probability.] P(n, k) is the number of ways to pick k objects out of a set of n objects. Again, the objects are supposed to be distinguishable. The formula is P(n, k) = n·(n–1)·(n–2)·…·(n–k+1) = n!/(n–k)!. That’s easy to understand intuitively: on your first pick you have n choices; on your second, n–1; on your third, n–2, etcetera. When n = k, we obviously get n! again.
There is a third concept: the k-combination (as opposed to the k-permutation), which we’ll denote by C(n, k). That’s when the order within our subset doesn’t matter: an ace, a queen and a jack taken out of some card deck are a queen, a jack, and an ace: we don’t care about the order. If we have k objects, there are k! ways of ordering them and, hence, we just have to divide P(n, k) by k! to get C(n, k). So we write: C(n, k) = P(n, k)/k! = n!/[(n–k)!k!]. You recognize C(n, k): it’s the binomial coeficient.
Now, the HyperPhysics example illustrating the three mentioned distributions (Maxwell-Boltzmann, Bose-Einstein and Fermi-Dirac) is a bit more complicated: we need to associate q energy levels with N particles. Every possible configuration is referred to as a micro-state, and the total number of possible micro-states is referred to as the multiplicity of the system, denoted by Ω(N, q). The formula for Ω(N, q) is another binomial coefficient: Ω(N, q) = (q+N–1)!/[q!(N–1)!]. Ω(N, q) = Ω(6, 9) = (9+6–1)!/[9!(6–1)!] = 2002.
In our example, however, we do not have distinct particles and, therefore, we only have 26 macro-states (as opposed to 2002 micro-states), which are also referred to, confusingly, as distributions or combinations.
Now, the number of micro-states associated with the same macro-state is given by yet another formula: it is equal to N!/[n1!·n2!·n3!·…·nq!], with ni! the number of particles in level i. [See why we need the 0! = 1 definition? It ensures unoccupied states do not affect the calculation.] So that’s how we get those numbers 6, 60 and 180 for those three macro-states.
But how do we calculate those average numbers of particles for each energy level? In other words, how do we calculate the probability densities under the Maxwell-Boltzmann, Fermi-Dirac and Bose-Einstein hypothesis respectively?
For the Maxwell-Boltzmann distribution, we proceed as follows: for each energy level j (or Ej, I should say), we calculate nj = ∑nij·Pi over all macro-states i. In this summation, we have nij, which is the number of particles in energy level j in micro-state i, while Pi is the probability of macro-state i as calculated by the ratio of (i) the number of micro-states associated with macro-state i and (ii) the total number of micro-states. For Pi, we gave the example of 3/2002 ≈ 0.3%. For 60 and 180, we get 60/2002 ≈ 3% and 180/2002 ≈ 9%. Calculating all the nj‘s for j ranging from 1 to 9 should yield the numbers and the graph below indeed.
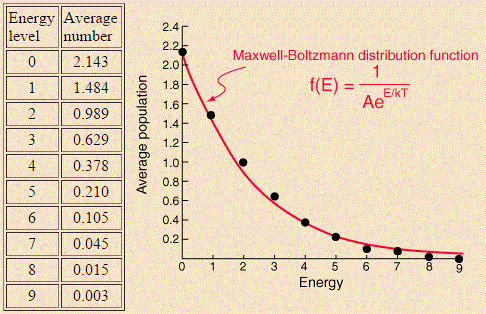 OK. That’s how it works for Maxwell-Boltzmann. Now, it is obvious that the Fermi-Dirac and the Bose-Einstein distribution should not be calculated in the same way because, if they were, they would not be different from the Maxwell-Boltzmann distribution! The trick is as follows.
OK. That’s how it works for Maxwell-Boltzmann. Now, it is obvious that the Fermi-Dirac and the Bose-Einstein distribution should not be calculated in the same way because, if they were, they would not be different from the Maxwell-Boltzmann distribution! The trick is as follows.
For the Bose-Einstein distribution, we give all macro-states equal weight—so that’s a weight of one, as shown below. Hence, the probability Pi is, quite simply, 1/26 ≈ 3.85% for all 26 macro-states. So we use the same nj = ∑nij·Pi formula but with Pi = 1/26.
Finally, I already explained how we get the Fermi-Dirac distribution: we can only have (i) one, (ii) two, or (iii) zero fermions for each energy level—not more than two! Hence, out of the 26 macro-states, only five are actually possible under the Fermi-Dirac hypothesis, as illustrated below once more. So it’s a very different distribution indeed!
Now, you’ll probably still have questions. For example, why does the assumption, for the Bose-Einstein analysis, that macro-states have equal probability favor the lower energy states? The answer is that the model also integrates other constraints: first, when associating a particle with an energy level, we do not favor one energy level over another, so all energy levels have equal probability. However, at the same time, the whole system has some fixed energy level, and so we cannot put the particles in the higher energy levels only! At the same time, we know that, if we have q particles, and the probability of a particle having some energy level j is the same for all j, then they are likely not to be all at the same energy level: they’ll be distributed, effectively, as evidenced by the very low chance (0.3% only) of having 5 particles in the ground state and 1 particle at a higher level, as opposed to the 3% and 9% chance of the other two combinations shown in that diagram with three possible Maxwell-Boltzmann (MB) combinations.
So what happens when assigning an equal probability to all 26 possible combinations (with value 1/26) is that the combinations that were previously rather unlikely – because they did have a rather heavy concentration of particles in the ground state only – are now much more likely. So that’s why the Bose-Einstein distribution, in this example at least, is skewed towards the lowest energy level—as compared to the Maxwell-Boltzmann distribution, that is.
So that’s what’s behind, and that should also answer the other question you surely have when looking at those five acceptable Fermi-Dirac configurations: why don’t we have the same five configurations starting from the top down, rather than from the bottom up? Now you know: such configuration would have much higher energy overall, and so that’s not allowed under this particular model.
There’s also this other question: we said the particles were indistinguishable, but so then we suddenly say there can be two at any energy level, because their spin is opposite. It’s obvious this is rather ad hoc as well. However, if we’d allow only one particle at any energy level, we’d have no allowable combinations and, hence, we’d have no Fermi-Dirac distribution at all in this example.
In short, the example is rather intuitive, which is actually why I like it so much: it shows how bosonic and fermionic behavior appear rather gradually, as a consequence of variables that are defined at the system level, such as density, or temperature. So, yes, you’re right if you think the HyperPhysics example lacks rigor. That’s why I think it’s such wonderful pedagogic device. 🙂