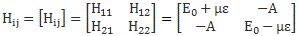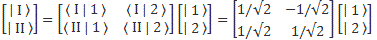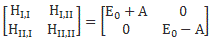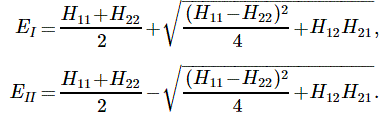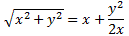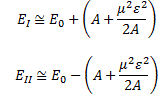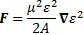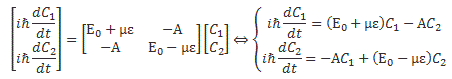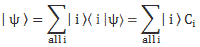Pre-script (dated 26 June 2020): I have come to the conclusion one does not need all this hocus-pocus to explain masers or lasers (and two-state systems in general): classical physics will do. So no use to read this. Read my papers instead. 🙂
Original post:
As I skipped the mathematical arguments in my previous post so as to focus on the essential results only, I thought it would be good to complement that post by looking at the math once again, so as to ensure we understand what it is that we’re doing. So let’s do that now. We start with the easy situation: free space.
The two-state system in free space
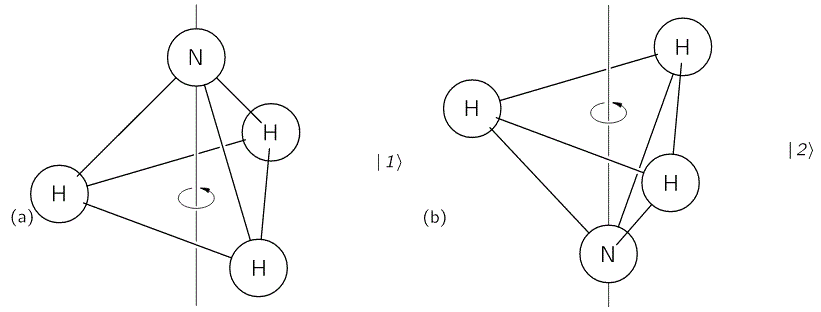
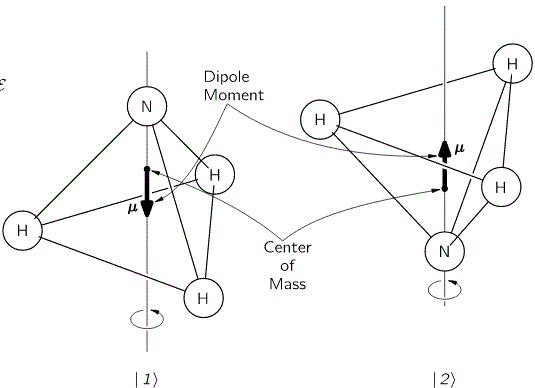
We started with an ammonia molecule in free space, i.e. we assumed there were no external force fields, like a gravitational or an electromagnetic force field. Hence, the picture was as simple as the one below: the nitrogen atom could be ‘up’ or ‘down’ with regard to its spin around its axis of symmetry.

It’s important to note that this ‘up’ or ‘down’ direction is defined in regard to the molecule itself, i.e. not in regard to some external reference frame. In other words, the reference frame is that of the molecule itself. For example, if I flip the illustration above – like below – then we’re still talking the same states, i.e. the molecule is still in state 1 in the image on the left-hand side and it’s still in state 2 in the image on the right-hand side.

We then modeled the uncertainty about its state by associating two different energy levels with the molecule: E0 + A and E0 − A. The idea is that the nitrogen atom needs to tunnel through a potential barrier to get to the other side of the plane of the hydrogens, and that requires energy. At the same time, we’ll show the two energy levels are effectively associated with an ‘up’ or ‘down’ direction of the electric dipole moment of the molecule. So that resembles the two spin states of an electron, which we associated with the +ħ/2 and −ħ/2 energies respectively. So if E0 would be zero (we can always take another reference point, remember?), then we’ve got the same thing: two energy levels that are separated by some definite amount: that amount is 2A for the ammonia molecule, and ħ when we’re talking quantum-mechanical spin. I should make a last note here, before I move on: note that these energies only make sense in the presence of some external field, because the + and − signs in the E0 + A and E0 − A and +ħ/2 and −ħ/2 expressions make sense only with regard to some external direction defining what’s ‘up’ and what’s ‘down’ really. But I am getting ahead of myself here. Let’s go back to free space: no external fields, so what’s ‘up’ or ‘down’ is completely random here. 🙂
Now, we also know an energy level can be associated with a complex-valued wavefunction, or an amplitude as we call it. To be precise, we can associate it with the generic a·e−(i/ħ)·(E·t − p∙x) expression which you know so well by now. Of course, as the reference frame is that of the molecule itself, its momentum is zero, so the p∙x term in the a·e−(i/ħ)·(E·t − p∙x) expression vanishes and the wavefunction reduces to a·e−i·ω·t = a·e−(i/ħ)·E·t, with ω = E/ħ. In other words, the energy level determines the temporal frequency, or the temporal variation (as opposed to the spatial frequency or variation), of the amplitude.
We then had to find the amplitudes C1(t) = 〈 1 | ψ 〉 and C2(t) =〈 2 | ψ 〉, so that’s the amplitude to be in state 1 or state 2 respectively. In my post on the Hamiltonian, I explained why the dynamics of a situation like this can be represented by the following set of differential equations:
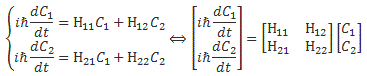
As mentioned, the C1 and C2 functions evolve in time, and so we should write them as C1 = C1(t) and C2 = C2(t) respectively. In fact, our Hamiltonian coefficients may also evolve in time, which is why it may be very difficult to solve those differential equations! However, as I’ll show below, one usually assumes they are constant, and then one makes informed guesses about them so as to find a solution that makes sense.
Now, I should remind you here of something you surely know: if C1 and C2 are solutions to this set of differential equations, then the superposition principle tells us that any linear combination a·C1 + b·C2 will also be a solution. So we need one or more extra conditions, usually some starting condition, which we can combine with a normalization condition, so we can get some unique solution that makes sense.
The Hij coefficients are referred to as Hamiltonian coefficients and, as shown in the mentioned post, the H11 and H22 coefficients are related to the amplitude of the molecule staying in state 1 and state 2 respectively, while the H12 and H21 coefficients are related to the amplitude of the molecule going from state 1 to state 2 and vice versa. Because of the perfect symmetry of the situation here, it’s easy to see that H11 should equal H22 , and that H12 and H21 should also be equal to each other. Indeed, Nature doesn’t care what we call state 1 or 2 here: as mentioned above, we did not define the ‘up’ and ‘down’ direction with respect to some external direction in space, so the molecule can have any orientation and, hence, switching the i an j indices should not make any difference. So that’s one clue, at least, that we can use to solve those equations: the perfect symmetry of the situation and, hence, the perfect symmetry of the Hamiltonian coefficients—in this case, at least!
The other clue is to think about the solution if we’d not have two states but one state only. In that case, we’d need to solve iħ·[dC1(t)/dt] = H11·C1(t). That’s simple enough, because you’ll remember that the exponential function is its own derivative. To be precise, we write: d(a·eiωt)/dt = a·d(eiωt)/dt = a·iω·eiωt, and please note that a can be any complex number: we’re not necessarily talking a real number here! In fact, we’re likely to talk complex coefficients, and we multiply with some other complex number (iω) anyway here! So if we write iħ·[dC1/dt] = H11·C1 as dC1/dt = −(i/ħ)·H11·C1 (remember: i−1 = 1/i = −i), then it’s easy to see that the C1 = a·e–(i/ħ)·H11·t function is the general solution for this differential equation. Let me write it out for you, just to make sure:
dC1/dt = d[a·e–(i/ħ)H11t]/dt = a·d[e–(i/ħ)H11t]/dt = –a·(i/ħ)·H11·e–(i/ħ)H11t
= –(i/ħ)·H11·a·e–(i/ħ)H11t = −(i/ħ)·H11·C1
Of course, that reminds us of our generic wavefunction a·e−(i/ħ)·E0·t wavefunction: we only need to equate H11 with E0 and we’re done! Hence, in a one-state system, the Hamiltonian coefficient is, quite simply, equal to the energy of the system. In fact, that’s a result can be generalized, as we’ll see below, and so that’s why Feynman says the Hamiltonian ought to be called the energy matrix.
In fact, we actually may have two states that are entirely uncoupled, i.e. a system in which there is no dependence of C1 on C2 and vice versa. In that case, the two equations reduce to:
iħ·[dC1/dt] = H11·C1 and iħ·[dC2/dt] = H22·C2
These do not form a coupled system and, hence, their solutions are independent:
C1(t) = a·e–(i/ħ)·H11·t and C2(t) = b·e–(i/ħ)·H22·t
The symmetry of the situation suggests we should equate a and b, and then the normalization condition says that the probabilities have to add up to one, so |C1(t)|2 + |C2(t)|2 = 1, so we’ll find that a = b = 1/√2.
OK. That’s simple enough, and this story has become quite long, so we should wrap it up. The two ‘clues’ – about symmetry and about the Hamiltonian coefficients being energy levels – lead Feynman to suggest that the Hamiltonian matrix for this particular case should be equal to:

Why? Well… It’s just one of Feynman’s clever guesses, and it yields probability functions that makes sense, i.e. they actually describe something real. That’s all. 🙂 I am only half-joking, because it’s a trial-and-error process indeed and, as I’ll explain in a separate section in this post, one needs to be aware of the various approximations involved when doing this stuff. So let’s be explicit about the reasoning here:
- We know that H11 = H22 = E0 if the two states would be identical. In other words, if we’d have only one state, rather than two – i.e. if H12 and H21 would be zero – then we’d just plug that in. So that’s what Feynman does. So that’s what we do here too! 🙂
- However, H12 and H21 are not zero, of course, and so assume there’s some amplitude to go from one position to the other by tunneling through the energy barrier and flipping to the other side. Now, we need to assign some value to that amplitude and so we’ll just assume that the energy that’s needed for the nitrogen atom to tunnel through the energy barrier and flip to the other side is equal to A. So we equate H12 and H21 with −A.
Of course, you’ll wonder: why minus A? Why wouldn’t we try H12 = H21 = A? Well… I could say that a particle usually loses potential energy as it moves from one place to another, but… Well… Think about it. Once it’s through, it’s through, isn’t it? And so then the energy is just E0 again. Indeed, if there’s no external field, the + or − sign is quite arbitrary. So what do we choose? The answer is: when considering our molecule in free space, it doesn’t matter. Using +A or −A yields the same probabilities. Indeed, let me give you the amplitudes we get for H11 = H22 = E0 and H12 and H21 = −A:
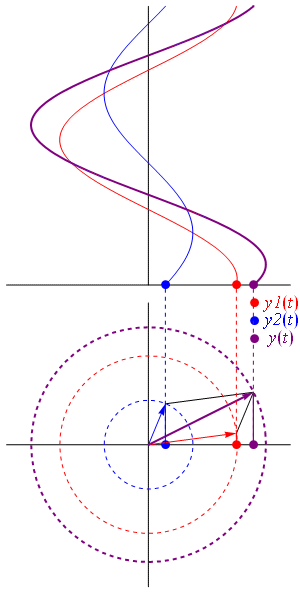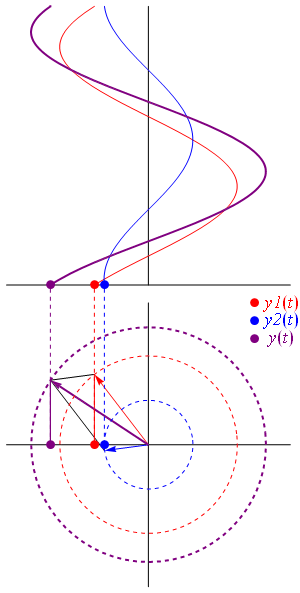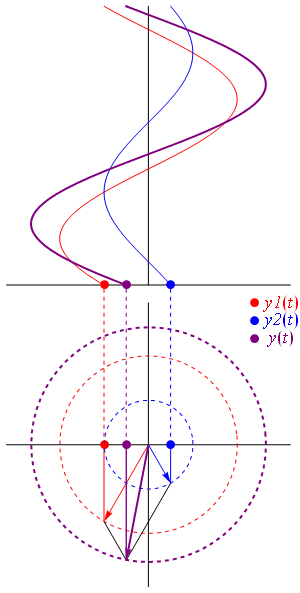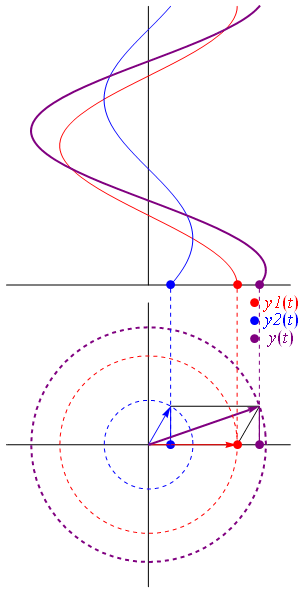
- C1(t) = 〈 1 | ψ 〉 = (1/2)·e−(i/ħ)·(E0 − A)·t + (1/2)·e−(i/ħ)·(E0 + A)·t = e−(i/ħ)·E0·t·cos[(A/ħ)·t]
- C2(t) = 〈 2 | ψ 〉 = (1/2)·e−(i/ħ)·(E0 − A)·t – (1/2)·e−(i/ħ)·(E0 + A)·t = i·e−(i/ħ)·E0·t·sin[(A/ħ)·t]
[In case you wonder how we go from those exponentials to a simple sine and cosine factor, remember that the sum of complex conjugates, i.e eiθ + e−iθ reduces to 2·cosθ, while eiθ − e−iθ reduces to 2·i·sinθ.]
Now, it’s easy to see that, if we’d have used +A rather than −A, we would have gotten something very similar:
- C1(t) = 〈 1 | ψ 〉 = (1/2)·e−(i/ħ)·(E0 + A)·t + (1/2)·e−(i/ħ)·(E0 − A)·t = e−(i/ħ)·E0·t·cos[(A/ħ)·t]
- C2(t) = 〈 2 | ψ 〉 = (1/2)·e−(i/ħ)·(E0 + A)·t – (1/2)·e−(i/ħ)·(E0 − A)·t = −i·e−(i/ħ)·E0·t·sin[(A/ħ)·t]
So we get a minus sign in front of our C2(t) function, because cos(α) = cos(–α) but sin(α) = −sin(α). However, the associated probabilities are exactly the same. For both, we get the same P1(t) and P2(t) functions:
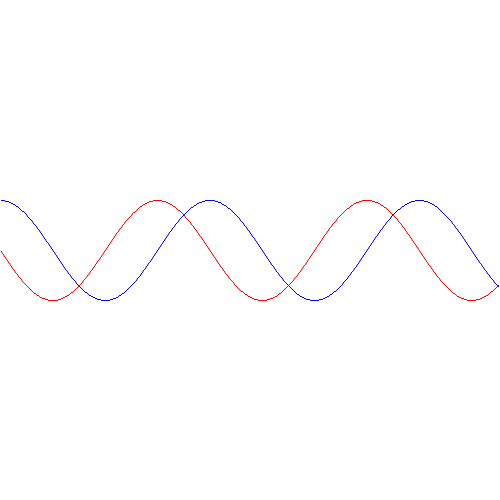
- P1(t) = |C1(t)|2 = cos2[(A/ħ)·t]
- P2(t) = |C2(t)|2 = sin2[(A/ħ)·t]
[Remember: the absolute square of i and −i is |i|2 = +√12 = +1 and |−i|2 = (−1)2|i|2 = +1 respectively, so the i and −i in the two C2(t) formulas disappear.]
You’ll remember the graph:

Of course, you’ll say: that plus or minus sign in front of C2(t) should matter somehow, doesn’t it? Well… Think about it. Taking the absolute square of some complex number – or some complex function , in this case! – amounts to multiplying it with its complex conjugate. Because the complex conjugate of a product is the product of the complex conjugates, it’s easy to see what happens: the e−(i/ħ)·E0·t factor in C1(t) = e−(i/ħ)·E0·t·cos[(A/ħ)·t] and C2(t) = ±i·e−(i/ħ)·E0·t·sin[(A/ħ)·t] gets multiplied by e+(i/ħ)·E0·t and, hence, doesn’t matter: e−(i/ħ)·E0·t·e+(i/ħ)·E0·t = e0 = 1. The cosine factor in C1(t) = e−(i/ħ)·E0·t·cos[(A/ħ)·t] is real, and so its complex conjugate is the same. Now, the ±i·sin[(A/ħ)·t] factor in C2(t) = ±i·e−(i/ħ)·E0·t·sin[(A/ħ)·t] is a pure imaginary number, and so its complex conjugate is its opposite. For some reason, we’ll find similar solutions for all of the situations we’ll describe below: the factor determining the probability will either be real or, else, a pure imaginary number. Hence, from a math point of view, it really doesn’t matter if we take +A or −A for or real factor for those H12 and H21 coefficients. We just need to be consistent in our choice, and I must assume that, in order to be consistent, Feynman likes to think of our nitrogen atom borrowing some energy from the system and, hence, temporarily reducing its energy by an amount that’s equal to −A. If you have a better interpretation, please do let me know! 🙂
OK. We’re done with this section… Except… Well… I have to show you how we got those C1(t) and C1(t) functions, no? Let me copy Feynman here:
 Note that the ‘trick’ involving the addition and subtraction of the differential equations is a trick we’ll use quite often, so please do have a look at it. As for the value of the a and b coefficients – which, as you can see, we’ve equated to 1 in our solutions for C1(t) and C1(t) – we get those because of the following starting condition: we assume that at t = 0, the molecule will be in state 1. Hence, we assume C1(0) = 1 and C2(0) = 0. In other words: we assume that we start out on that P1(t) curve in that graph with the probability functions above, so the C1(0) = 1 and C2(0) = 0 starting condition is equivalent to P1(0) = 1 and P1(0) = 0. Plugging that in gives us a/2 + b/2 = 1 and a/2 − b/2 = 0, which is possible only if a = b = 1.
Note that the ‘trick’ involving the addition and subtraction of the differential equations is a trick we’ll use quite often, so please do have a look at it. As for the value of the a and b coefficients – which, as you can see, we’ve equated to 1 in our solutions for C1(t) and C1(t) – we get those because of the following starting condition: we assume that at t = 0, the molecule will be in state 1. Hence, we assume C1(0) = 1 and C2(0) = 0. In other words: we assume that we start out on that P1(t) curve in that graph with the probability functions above, so the C1(0) = 1 and C2(0) = 0 starting condition is equivalent to P1(0) = 1 and P1(0) = 0. Plugging that in gives us a/2 + b/2 = 1 and a/2 − b/2 = 0, which is possible only if a = b = 1.
Of course, you’ll say: what if we’d choose to start out with state 2, so our starting condition is P1(0) = 0 and P1(0) = 1? Then a = 1 and b = −1, and we get the solution we got when equating H12 and H21 with +A, rather than with −A. So you can think about that symmetry once again: when we’re in free space, then it’s quite arbitrary what we call ‘up’ or ‘down’.
So… Well… That’s all great. I should, perhaps, just add one more note, and that’s on that A/ħ value. We calculated it in the previous post, because we wanted to actually calculate the period of those P1(t) and P2(t) functions. Because we’re talking the square of a cosine and a sine respectively, the period is equal to π, rather than 2π, so we wrote: (A/ħ)·T = π ⇔ T = π·ħ/A. Now, the separation between the two energy levels E0 + A and E0 − A, so that’s 2A, has been measured as being equal, more or less, to 2A ≈ 10−4 eV.
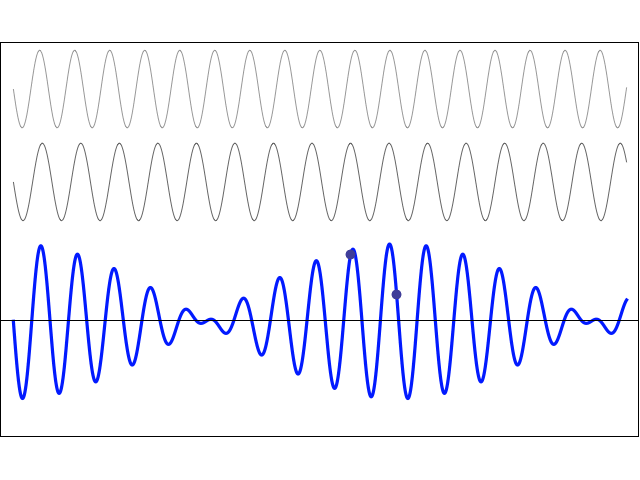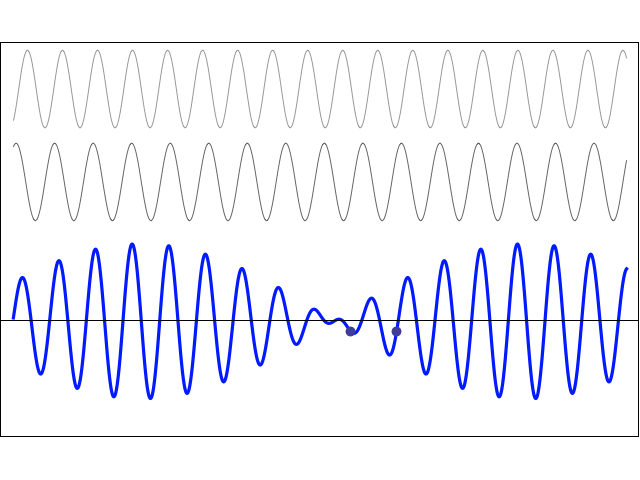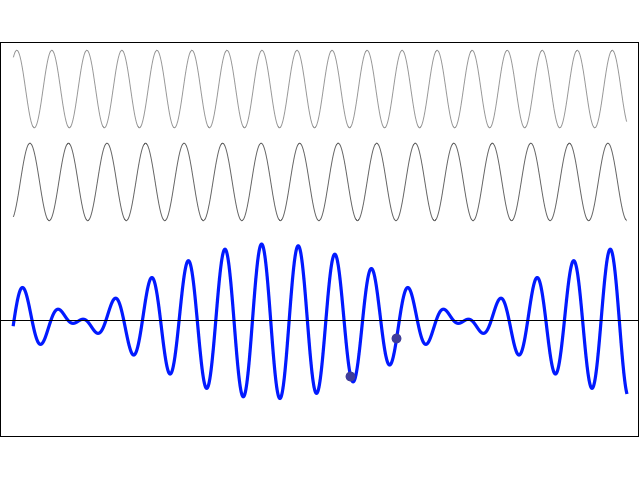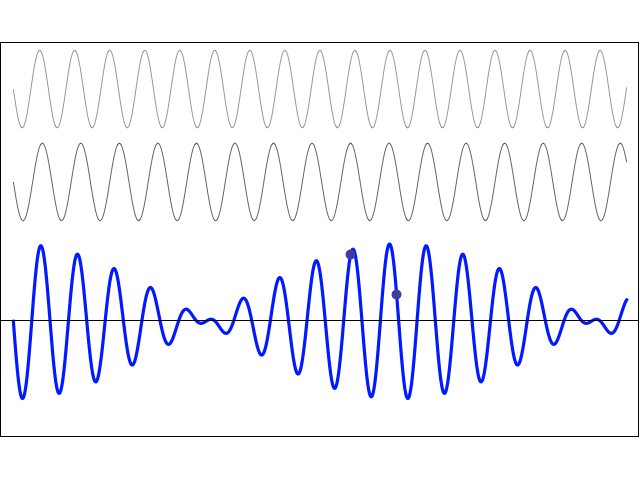
How does one measure that? As mentioned above, I’ll show you, in a moment, that, when applying some external field, the plus and minus sign do matter, and the separation between those two energy levels E0 + A and E0 − A will effectively represent something physical. More in particular, we’ll have transitions from one energy level to another and that corresponds to electromagnetic radiation being emitted or absorbed, and so there’s a relation between the energy and the frequency of that radiation. To be precise, we can write 2A = h·f0. The frequency of the radiation that’s being absorbed or emitted is 23.79 GHz, which corresponds to microwave radiation with a wavelength of λ = c/f0 = 1.26 cm. Hence, 2·A ≈ 25×109 Hz times 4×10−15 eV·s = 10−4 eV, indeed, and, therefore, we can write: T = π·ħ/A ≈ 3.14 × 6.6×10−16 eV·s divided by 0.5×10−4 eV, so that’s 40×10−12 seconds = 40 picoseconds. That’s 40 trillionths of a seconds. So that’s very short, and surely much shorter than the time that’s associated with, say, a freely emitting sodium atom, which is of the order of 3.2×10−8 seconds. You may think that makes sense, because the photon energy is so much lower: a sodium light photon is associated with an energy equal to E = h·f = 500×1012 Hz times 4×10−15 eV·s = 2 eV, so that’s 20,000 times 10−4 eV.
There’s a funny thing, however. An oscillation of a frequency of 500 tera-hertz that lasts 3.2×10−8 seconds is equivalent to 500×1012 Hz times 3.2×10−8 s ≈ 16 million cycles. However, an oscillation of a frequency of 23.97 giga-hertz that only lasts 40×10−12 seconds is equivalent to 23.97×109 Hz times 40×10−12 s ≈ 1000×10−3 = 1 ! One cycle only? We’re surely not talking resonance here!
So… Well… I am just flagging it here. We’ll have to do some more thinking about that later. [I’ve added an addendum that may or may not help us in this regard. :-)]
The two-state system in a field
As mentioned above, when there is no external force field, we define the ‘up’ or ‘down’ direction of the nitrogen atom was defined with regard to its its spin around its axis of symmetry, so with regard to the molecule itself. However, when we apply an external electromagnetic field, as shown below, we do have some external reference frame.
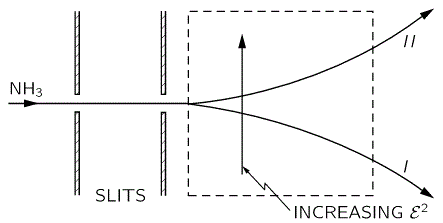
Now, the external reference frame – i.e. the physics of the situation, really – may make it more convenient to define the whole system using another set of base states, which we’ll refer to as I and II, rather than 1 and 2. Indeed, you’ve seen the picture below: it shows a state selector, or a filter as we called it. In this case, there’s a filtering according to whether our ammonia molecule is in state I or, alternatively, state II. It’s like a Stern-Gerlach apparatus splitting an electron beam according to the spin state of the electrons, which is ‘up’ or ‘down’ too, but in a totally different way than our ammonia molecule. Indeed, the ‘up’ and ‘down’ spin of an electron has to do with its magnetic moment and its angular momentum. However, there are a lot of similarities here, and so you may want to compare the two situations indeed, i.e. the electron beam in an inhomogeneous magnetic field versus the ammonia beam in an inhomogeneous electric field.

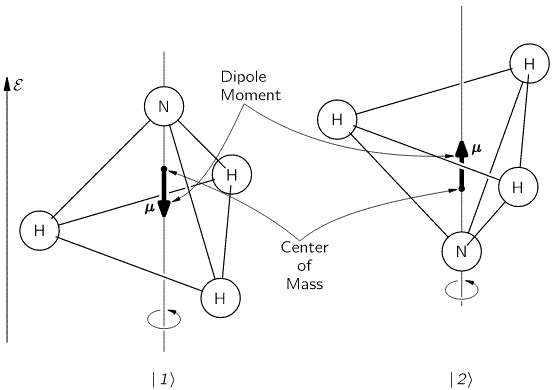
Now, when reading Feynman, as he walks us through the relevant Lecture on all of this, you get the impression that it’s the I and II states only that have some kind of physical or geometric interpretation. That’s not the case. Of course, the diagram of the state selector above makes it very obvious that these new I and II base states make very much sense in regard to the orientation of the field, i.e. with regard to external space, rather than with respect to the position of our nitrogen atom vis-á-vis the hydrogens. But… Well… Look at the image below: the direction of the field (which we denote by ε because we’ve been using the E for energy) obviously matters when defining the old ‘up’ and ‘down’ states of our nitrogen atom too!
In other words, our previous | 1 〉 and | 2 〉 base states acquire a new meaning too: it obviously matters whether or not the electric dipole moment of the molecule is in the same or, conversely, in the opposite direction of the field. To be precise, the presence of the electromagnetic field suddenly gives the energy levels that we’d associate with these two states a very different physical interpretation.

Indeed, from the illustration above, it’s easy to see that the electric dipole moment of this particular molecule in state 1 is in the opposite direction and, therefore, temporarily ignoring the amplitude to flip over (so we do not think of A for just a brief little moment), the energy that we’d associate with state 1 would be equal to E0 + με. Likewise, the energy we’d associate with state 2 is equal to E0 − με. Indeed, you’ll remember that the (potential) energy of an electric dipole is equal to the vector dot product of the electric dipole moment μ and the field vector ε, but with a minus sign in front so as to get the sign for the energy righ. So the energy is equal to −μ·ε = −|μ|·|ε|·cosθ, with θ the angle between both vectors. Now, the illustration above makes it clear that state 1 and 2 are defined for θ = π and θ = 0 respectively. [And, yes! Please do note that state 1 is the highest energy level, because it’s associated with the highest potential energy: the electric dipole moment μ of our ammonia molecule will – obviously! – want to align itself with the electric field ε ! Just think of what it would imply to turn the molecule in the field!]
Therefore, using the same hunches as the ones we used in the free space example, Feynman suggests that, when some external electric field is involved, we should use the following Hamiltonian matrix:

So we’ll need to solve a similar set of differential equations with this Hamiltonian now. We’ll do that later and, as mentioned above, it will be more convenient to switch to another set of base states, or another ‘representation’ as it’s referred to. But… Well… Let’s not get too much ahead of ourselves: I’ll say something about that before we’ll start solving the thing, but let’s first look at that Hamiltonian once more.
When I say that Feynman uses the same clues here, then… Well.. That’s true and not true. You should note that the diagonal elements in the Hamiltonian above are not the same: E0 + με ≠ E0 + με. So we’ve lost that symmetry of free space which, from a math point of view, was reflected in those identical H11 = H22 = E0 coefficients.
That should be obvious from what I write above: state 1 and state 2 are no longer those 1 and 2 states we described when looking at the molecule in free space. Indeed, the | 1 〉 and | 2 〉 states are still ‘up’ or ‘down’, but the illustration above also makes it clear we’re defining state 1 and state 2 not only with respect to the molecule’s spin around its own axis of symmetry but also vis-á-vis some direction in space. To be precise, we’re defining state 1 and state 2 here with respect to the direction of the electric field ε. Now that makes a really big difference in terms of interpreting what’s going on.
In fact, the ‘splitting’ of the energy levels because of that amplitude A is now something physical too, i.e. something that goes beyond just modeling the uncertainty involved. In fact, we’ll find it convenient to distinguish two new energy levels, which we’ll write as EI = E0 + A and EII = E0 − A respectively. They are, of course, related to those new base states | I 〉 and | II 〉 that we’ll want to use. So the E0 + A and E0 − A energy levels themselves will acquire some physical meaning, and especially the separation between them, i.e. the value of 2A. Indeed, EI = E0 + A and EII = E0 − A will effectively represent an ‘upper’ and a ‘lower’ energy level respectively.
But, again, I am getting ahead of myself. Let’s first, as part of working towards a solution for our equations, look at what happens if and when we’d switch to another representation indeed.
Switching to another representation
Let me remind you of what I wrote in my post on quantum math in this regard. The actual state of our ammonia molecule – or any quantum-mechanical system really – is always to be described in terms of a set of base states. For example, if we have two possible base states only, we’ll write:
| φ 〉 = | 1 〉 C1 + | 2 〉 C2
You’ll say: why? Our molecule is obviously always in either state 1 or state 2, isn’t it? Well… Yes and no. That’s the mystery of quantum mechanics: it is and it isn’t. As long as we don’t measure it, there is an amplitude for it to be in state 1 and an amplitude for it to be in state 2. So we can only make sense of its state by actually calculating 〈 1 | φ 〉 and 〈 2 | φ 〉 which, unsurprisingly are equal to 〈 1 | φ 〉 = 〈 1 | 1 〉 C1 + 〈 1 | 2 〉 C2 = C1(t) and 〈 2 | φ 〉 = 〈 2 | 1 〉 C1 + 〈 2 | 2 〉 C2 = C2(t) respectively, and so these two functions give us the probabilities P1(t) and P2(t) respectively. So that’s Schrödinger’s cat really: the cat is dead or alive, but we don’t know until we open the box, and we only have a probability function – so we can say that it’s probably dead or probably alive, depending on the odds – as long as we do not open the box. It’s as simple as that.
Now, the ‘dead’ and ‘alive’ condition are, obviously, the ‘base states’ in Schrödinger’s rather famous example, and we can write them as | DEAD 〉 and | ALIVE 〉 you’d agree it would be difficult to find another representation. For example, it doesn’t make much sense to say that we’ve rotated the two base states over 90 degrees and we now have two new states equal to (1/√2)·| DEAD 〉 – (1/√2)·| ALIVE 〉 and (1/√2)·| DEAD 〉 + (1/√2)·| ALIVE 〉 respectively. There’s no direction in space in regard to which we’re defining those two base states: dead is dead, and alive is alive.
The situation really resembles our ammonia molecule in free space: there’s no external reference against which to define the base states. However, as soon as some external field is involved, we do have a direction in space and, as mentioned above, our base states are now defined with respect to a particular orientation in space. That implies two things. The first is that we should no longer say that our molecule will always be in either state 1 or state 2. There’s no reason for it to be perfectly aligned with or against the field. Its orientation can be anything really, and so its state is likely to be some combination of those two pure base states | 1 〉 and | 2 〉.
The second thing is that we may choose another set of base states, and specify the very same state in terms of the new base states. So, assuming we choose some other set of base states | I 〉 and | II 〉, we can write the very same state | φ 〉 = | 1 〉 C1 + | 2 〉 C2 as:
| φ 〉 = | I 〉 CI + | II 〉 CII
It’s really like what you learned about vectors in high school: one can go from one set of base vectors to another by a transformation, such as, for example, a rotation, or a translation. It’s just that, just like in high school, we need some direction in regard to which we define our rotation or our translation.
For state vectors, I showed how a rotation of base states worked in one of my posts on two-state systems. To be specific, we had the following relation between the two representations:
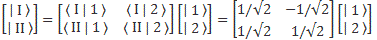
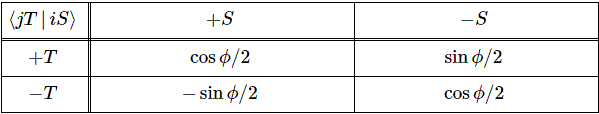
The (1/√2) factor is there because of the normalization condition, and the two-by-two matrix equals the transformation matrix for a rotation of a state filtering apparatus about the y-axis, over an angle equal to (minus) 90 degrees, which we wrote as:

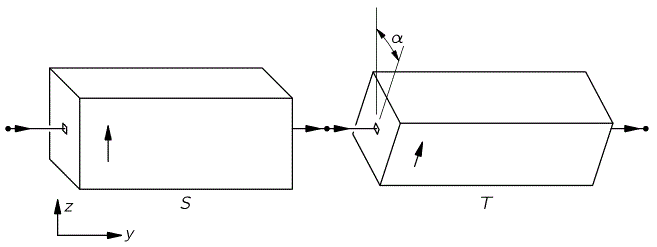
The y-axis? What y-axis? What state filtering apparatus? Just relax. Think about what you’ve learned already. The orientations are shown below: the S apparatus separates ‘up’ and ‘down’ states along the z-axis, while the T-apparatus does so along an axis that is tilted, about the y-axis, over an angle equal to α, or φ, as it’s written in the table above.

Of course, we don’t really introduce an apparatus at this or that angle. We just introduced an electromagnetic field, which re-defined our | 1 〉 and | 2 〉 base states and, therefore, through the rotational transformation matrix, also defines our | I 〉 and | II 〉 base states.
[…] You may have lost me by now, and so then you’ll want to skip to the next section. That’s fine. Just remember that the representations in terms of | I 〉 and | II 〉 base states or in terms of | 1 〉 and | 2 〉 base states are mathematically equivalent. Having said that, if you’re reading this post, and you want to understand it, truly (because you want to truly understand quantum mechanics), then you should try to stick with me here. 🙂 Indeed, there’s a zillion things you could think about right now, but you should stick to the math now. Using that transformation matrix, we can relate the CI and CII coefficients in the | φ 〉 = | I 〉 CI + | II 〉 CII expression to the CI and CII coefficients in the | φ 〉 = | 1 〉 C1 + | 2 〉 C2 expression. Indeed, we wrote:
- CI = 〈 I | ψ 〉 = (1/√2)·(C1 − C2)
- CII = 〈 II | ψ 〉 = (1/√2)·(C1 + C2)
That’s exactly the same as writing:

OK. […] Waw! You just took a huge leap, because we can now compare the two sets of differential equations:

They’re mathematically equivalent, but the mathematical behavior of the functions involved is very different. Indeed, unlike the C1(t) and C2(t) amplitudes, we find that the CI(t) and CII(t) amplitudes are stationary, i.e. the associated probabilities – which we find by taking the absolute square of the amplitudes, as usual – do not vary in time. To be precise, if you write it all out and simplify, you’ll find that the CI(t) and CII(t) amplitudes are equal to:
- CI(t) = 〈 I | ψ 〉 = (1/√2)·(C1 − C2) = (1/√2)·e−(i/ħ)·(E0+ A)·t = (1/√2)·e−(i/ħ)·EI·t
- CII(t) = 〈 II | ψ 〉 = (1/√2)·(C1 + C2) = (1/√2)·e−(i/ħ)·(E0− A)·t = (1/√2)·e−(i/ħ)·EII·t
As the absolute square of the exponential is equal to one, the associated probabilities, i.e. |CI(t)|2 and |CII(t)|2, are, quite simply, equal to |1/√2|2 = 1/2. Now, it is very tempting to say that this means that our ammonia molecule has an equal chance to be in state I or state II. In fact, while I may have said something like that in my previous posts, that’s not how one should interpret this. The chance of our molecule being exactly in state I or state II, or in state 1 or state 2 is varying with time, with the probability being ‘dumped’ from one state to the other all of the time.
I mean… The electric dipole moment can point in any direction, really. So saying that our molecule has a 50/50 chance of being in state 1 or state 2 makes no sense. Likewise, saying that our molecule has a 50/50 chance of being in state I or state II makes no sense either. Indeed, the state of our molecule is specified by the | φ 〉 = | I 〉 CI + | II 〉 CII = | 1 〉 C1 + | 2 〉 C2 equations, and neither of these two expressions is a stationary state. They mix two frequencies, because they mix two energy levels.
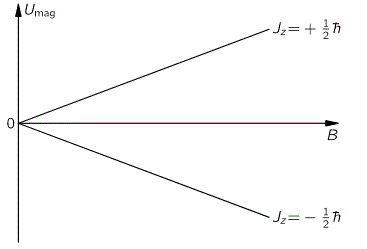
Having said that, we’re talking quantum mechanics here and, therefore, an external inhomogeneous electric field will effectively split the ammonia molecules according to their state. The situation is really like what a Stern-Gerlach apparatus does to a beam of electrons: it will split the beam according to the electron’s spin, which is either ‘up’ or, else, ‘down’, as shown in the graph below:

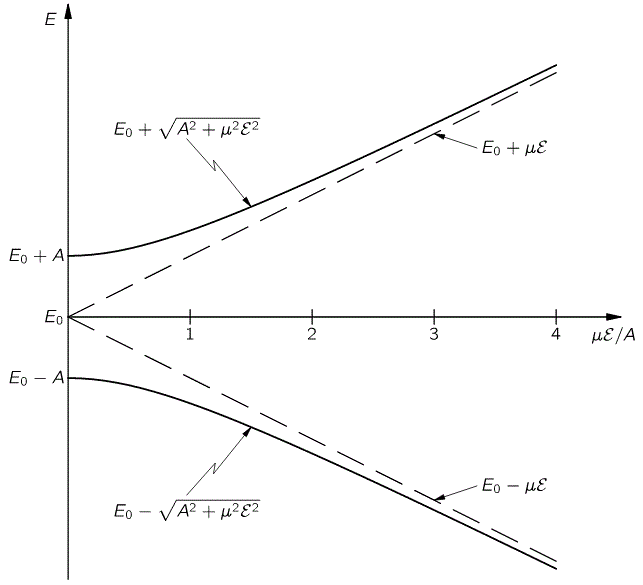
The graph for our ammonia molecule, shown below, is very similar. The vertical axis measures the same: energy. And the horizontal axis measures με, which increases with the strength of the electric field ε. So we see a similar ‘splitting’ of the energy of the molecule in an external electric field.

How should we explain this? It is very tempting to think that the presence of an external force field causes the electrons, or the ammonia molecule, to ‘snap into’ one of the two possible states, which are referred to as state I and state II respectively in the illustration of the ammonia state selector below. But… Well… Here we’re entering the murky waters of actually interpreting quantum mechanics, for which (a) we have no time, and (b) we are not qualified. So you should just believe, or take for granted, what’s being shown here: an inhomogeneous electric field will split our ammonia beam according to their state, which we define as I and II respectively, and which are associated with the energy E0+ A and E0− A respectively.
As mentioned above, you should note that these two states are stationary. The Hamiltonian equations which, as they always do, describe the dynamics of this system, imply that the amplitude to go from state I to state II, or vice versa, is zero. To make sure you ‘get’ that, I reproduce the associated Hamiltonian matrix once again:
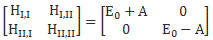
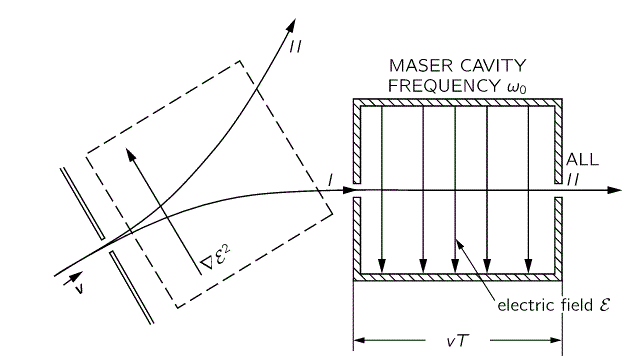
Of course, that will change when we start our analysis of what’s happening in the maser. Indeed, we will have some non-zero HI,II and HII,I amplitudes in the resonant cavity of our ammonia maser, in which we’ll have an oscillating electric field and, as a result, induced transitions from state I to II and vice versa. However, that’s for later. While I’ll quickly insert the full picture diagram below, you should, for the moment, just think about those two stationary states and those two zeroes. 🙂

Capito? If not… Well… Start reading this post again, I’d say. 🙂
Intermezzo: on approximations
At this point, I need to say a few things about all of the approximations involved, because it can be quite confusing indeed. So let’s take a closer look at those energy levels and the related Hamiltonian coefficients. In fact, in his Lectures, Feynman shows us that we can always have a general solution for the Hamiltonian equations describing a two-state system whenever we have constant Hamiltonian coefficients. That general solution – which, mind you, is derived assuming Hamiltonian coefficients that do not depend on time – can always be written in terms of two stationary base states, i.e. states with a definite energy and, hence, a constant probability. The equations, and the two definite energy levels are:


That yields the following values for the energy levels for the stationary states:
Now, that’s very different from the EI = E0+ A and EII = E0− A energy levels for those stationary states we had defined in the previous section: those stationary states had no square root, and no μ2ε2, in their energy. In fact, that sort of answers the question: if there’s no external field, then that μ2ε2 factor is zero, and the square root in the expression becomes ±√A2 = ±A. So then we’re back to our EI = E0+ A and EII = E0− A formulas. The whole point, however, is that we will actually have an electric field in that cavity. Moreover, it’s going to be a field that varies in time, which we’ll write:
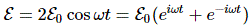
Now, part of the confusion in Feynman’s approach is that he constantly switches between representing the system in terms of the I and II base states and the 1 and 2 base states respectively. For a good understanding, we should compare with our original representation of the dynamics in free space, for which the Hamiltonian was the following one:
That matrix can easily be related to the new one we’re going to have to solve, which is equal to:
The interpretation is easy if we look at that illustration again:
If the direction of the electric dipole moment is opposite to the direction ε, then the associated energy is equal to −μ·ε = −μ·ε = −|μ|·|ε|·cosθ = −μ·ε·cos(π) = +με. Conversely, for state 2, we find −μ·ε·cos(0) = −με for the energy that’s associated with the dipole moment. You can and should think about the physics involved here, because they make sense! Thinking of amplitudes, you should note that the +με and −με terms effectively change the H11 and H22 coefficients, so they change the amplitude to stay in state 1 or state 2 respectively. That, of course, will have an impact on the associated probabilities, and so that’s why we’re talking of induced transitions now.
Having said that, the Hamiltonian matrix above keeps the −A for H12 and H21, so the matrix captures spontaneous transitions too!
Still… You may wonder why Feynman doesn’t use those EI and EII formulas with the square root because that would give us some exact solution, wouldn’t it? The answer to that question is: maybe it would, but would you know how to solve those equations? We’ll have a varying field, remember? So our Hamiltonian H11 and H22 coefficients will no longer be constant, but time-dependent. As you’re going to see, it takes Feynman three pages to solve the whole thing using the +με and −με approximation. So just imagine how complicated it would be using that square root expression! [By the way, do have a look at those asymptotic curves in that illustration showing the splitting of energy levels above, so you see how that approximation looks like.]
So that’s the real answer: we need to simplify somehow, so as to get any solutions at all!
Of course, it’s all quite confusing because, after Feynman first notes that, for strong fields, the A2 in that square root is small as compared to μ2ε2, thereby justifying the use of the simplified EI = E0+ με = H11 and EII = E0− με = H22 coefficients, he continues and bluntly uses the very same square root expression to explain how that state selector works, saying that the electric field in the state selector will be rather weak and, hence, that με will be much smaller than A, so one can use the following approximation for the square root in the expressions above:
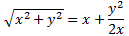 The energy expressions then reduce to:
The energy expressions then reduce to: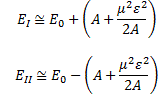
And then we can calculate the force on the molecules as:
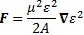
So the electric field in the state selector is weak, but the electric field in the cavity is supposed to be strong, and so… Well… That’s it, really. The bottom line is that we’ve a beam of ammonia molecules that are all in state I, and it’s what happens with that beam then, that is being described by our new set of differential equations:

Solving the equations
As all molecules in our ammonia beam are described in terms of the | I 〉 and | II 〉 base states – as evidenced by the fact that we say all molecules that enter the cavity are state I – we need to switch to that representation. We do that by using that transformation above, so we write:
- CI = 〈 I | ψ 〉 = (1/√2)·(C1 − C2)
- CII = 〈 II | ψ 〉 = (1/√2)·(C1 + C2)
Keeping these ‘definitions’ of CI and CII in mind, you should then add the two differential equations, divide the result by the square root of 2, and you should get the following new equation:
Please! Do it and verify the result! You want to learn something here, no? 🙂
Likewise, subtracting the two differential equations, we get:
We can re-write this as:
Now, the problem is that the Hamiltonian constants here are not constant. To be precise, the electric field ε varies in time. We wrote:
So HI,II and HII,I, which are equal to με, are not constant: we’ve got Hamiltonian coefficients that are a function of time themselves. […] So… Well… We just need to get on with it and try to finally solve this thing. Let me just copy Feynman as he grinds through this:
This is only the first step in the process. Feynman just takes two trial functions, which are really similar to the very general C1 = a·e–(i/ħ)·H11·t function we presented when only one equation was involved, or – if you prefer a set of two equations – those CI(t) = a·e−(i/ħ)·EI·t and CI(t) = b·e−(i/ħ)·EII·t equations above. The difference is that the coefficients in front, i.e. γI and γII are not some (complex) constant, but functions of time themselves. The next step in the derivation is as follows:
One needs to do a bit of gymnastics here as well to follow what’s going on, but please do check and you’ll see it works. Feynman derives another set of differential equations here, and they specify these γI = γI(t) and γII = γII(t) functions. These equations are written in terms of the frequency of the field, i.e. ω, and the resonant frequency ω0, which we mentioned above when calculating that 23.79 GHz frequency from the 2A = h·f0 equation. So ω0 is the same molecular resonance frequency but expressed as an angular frequency, so ω0 = f0/2π = ħ/2A. He then proceeds to simplify, using assumptions one should check. He then continues:
That gives us what we presented in the previous post:
So… Well… What to say? I explained those probability functions in my previous post, indeed. We’ve got two probabilities here:
- PI = cos2[(με0/ħ)·t]
- PII = sin2[(με0/ħ)·t]
So that’s just like the P1 = cos2[(A/ħ)·t] and P2 = sin2[(A/ħ)·t] probabilities we found for spontaneous transitions. But so here we are talking induced transitions.
As you can see, the frequency and, hence, the period, depend on the strength, or magnitude, of the electric field, i.e. the ε0 constant in the ε = 2ε0cos(ω·t) expression. The natural unit for measuring time would be the period once again, which we can easily calculate as (με0/ħ)·T = π ⇔ T = π·ħ/με0.
Now, we had that T = (π·ħ)/(2A) expression above, which allowed us to calculate the period of the spontaneous transition frequency, which we found was like 40 picoseconds, i.e. 40×10−12 seconds. Now, the T = (π·ħ)/(2με0) is very similar, it allows us to calculate the expected, average, or mean time for an induced transition. In fact, if we write Tinduced = (π·ħ)/(2με0) and Tspontaneous = (π·ħ)/(2A), then we can take ratio to find:
Tinduced/Tspontaneous = [(π·ħ)/(2με0)]/[(π·ħ)/(2A)] = A/με0
This A/με0 ratio is greater than one, so Tinduced/Tspontaneous is greater than one, which, in turn, means that the presence of our electric field – which, let me remind you, dances to the beat of the resonant frequency – causes a slower transition than we would have had if the oscillating electric field were not present.
But – Hey! – that’s the wrong comparison! Remember all molecules enter in a stationary state, as they’ve been selected so as to ensure they’re in state I. So there is no such thing as a spontaneous transition frequency here! They’re all polarized, so to speak, and they would remain that way if there was no field in the cavity. So if there was no oscillating electric field, they would never transition. Nothing would happen! Well… In terms of our particular set of base states, of course! Why? Well… Look at the Hamiltonian coefficients HI,II = HII,I = με: these coefficients are zero if ε is zero. So… Well… That says it all.
So that‘s what it’s all about: induced emission and, as I explained in my previous post, because all molecules enter in state I, i.e. the upper energy state, literally, they all ‘dump’ a net amount of energy equal to 2A into the cavity at the occasion of their first transition. The molecules then keep dancing, of course, and so they absorb and emit the same amount as they go through the cavity, but… Well… We’ve got a net contribution here, which is not only enough to maintain the cavity oscillations, but actually also provides a small excess of power that can be drawn from the cavity as microwave radiation of the same frequency.
As Feynman notes, an exact description of what actually happens requires an understanding of the quantum mechanics of the field in the cavity, i.e. quantum field theory, which I haven’t studied yet. But… Well… That’s for later, I guess. 🙂
Post scriptum: The sheer length of this post shows we’re not doing something that’s easy here. Frankly, I feel the whole analysis is still quite obscure, in the sense that – despite looking at this thing again and again – it’s hard to sort of interpret what’s going on, in a physical sense that is. But perhaps one shouldn’t try that. I’ve quoted Feynman’s view on how easy or how difficult it is to ‘understand’ quantum mechanics a couple of times already, so let me do it once more:
“Because atomic behavior is so unlike ordinary experience, it is very difficult to get used to, and it appears peculiar and mysterious to everyone—both to the novice and to the experienced physicist. Even the experts do not understand it the way they would like to, and it is perfectly reasonable that they should not, because all of direct, human experience and human intuition applies to large objects.”
So… Well… I’ll grind through the remaining Lectures now – I am halfway through Volume III now – and then re-visit all of this. Despite Feynman’s warning, I want to understand it the way I like to, even if I don’t quite know what way that is right now. 🙂
Addendum: As for those cycles and periods, I noted a couple of times already that the Planck-Einstein equation E = h·f can usefully be re-written as E/f = h, as it gives a physical interpretation to the value of the Planck constant. In fact, I said h is the energy that’s associated with one cycle, regardless of the frequency of the radiation involved. Indeed, the energy of a photon divided by the number of cycles per second, should give us the energy per cycle, no?
Well… Yes and no. Planck’s constant h and the frequency f are both expressed referencing the time unit. However, if we say that a sodium atom emits one photon only as its electron transitions from a higher energy level to a lower one, and if we say that involves a decay time of the order of 3.2×10−8 seconds, then what we’re saying really is that a sodium light photon will ‘pack’ like 16 million cycles, which is what we get when we multiply the number of cycles per second (i.e. the mentioned frequency of 500×1012 Hz) by the decay time (i.e. 3.2×10−8 seconds): (500×1012 Hz)·(3.2×10−8 s) = 16 ×106 cycles, indeed. So the energy per cycle is 2.068 eV (i.e. the photon energy) divided by 16×106, so that’s 0.129×10−6 eV. Unsurprisingly, that’s what we get when we we divide h by 3.2×10−8 s: (4.13567×10−15)/(3.2×10−8 s) = 1.29×10−7 eV. We’re just putting some values in to the E/(f·T) = h/T equation here.
The logic for that 2A = h·f0 is the same. The frequency of the radiation that’s being absorbed or emitted is 23.79 GHz, so the photon energy is (23.97×109 Hz)·(4.13567×10−15 eV·s) ≈ 1×10−4 eV. Now, we calculated the transition period T as T = π·ħ/A ≈ (π·6.626×10−16 eV·s)/(0.5×10−4 eV) ≈ 41.6×10−12 seconds. Now, an oscillation of a frequency of 23.97 giga-hertz that only lasts 41.6×10−12 seconds is an oscillation of one cycle only. The consequence is that, when we continue this style of reasoning, we’d have a photon that packs all of its energy into one cycle!
Let’s think about what this implies in terms of the density in space. The wavelength of our microwave radiation is 1.25×10−2 m, so we’ve got a ‘density’ of 1×10−4 eV/1.25×10−2 m = 0.8×10−2 eV/m = 0.008 eV/m. The wavelength of our sodium light is 0.6×10−6 m, so we get a ‘density’ of 1.29×10−7 eV/0.6×10−6 m = 2.15×10−1 eV/m = 0.215 eV/m. So the energy ‘density’ of our sodium light is 26.875 times that of our microwave radiation. 🙂
Frankly, I am not quite sure if calculations like this make much sense. In fact, when talking about energy densities, I should review my posts on the Poynting vector. However, they may help you think things through. 🙂
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 17, 2020 as a result of a DMCA takedown notice from Michael A. Gottlieb, Rudolf Pfeiffer, and The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/