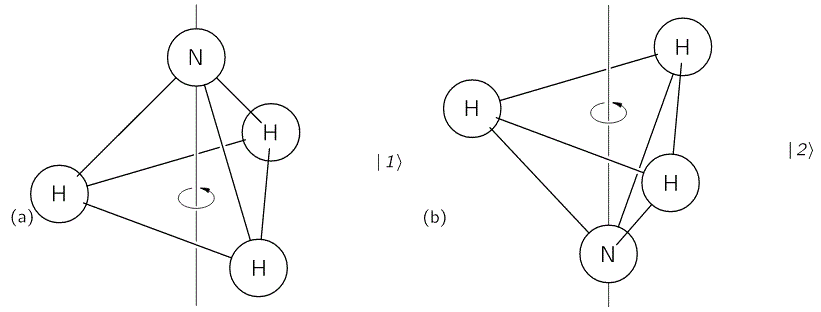
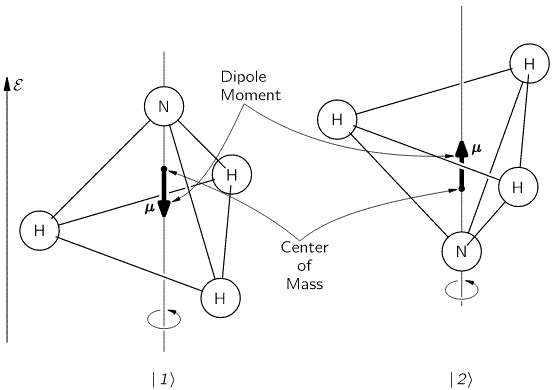
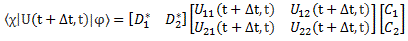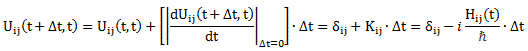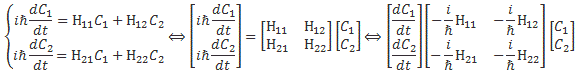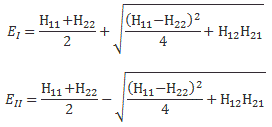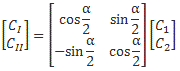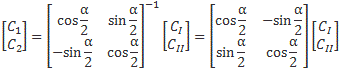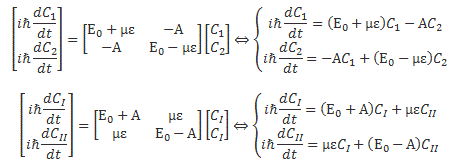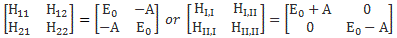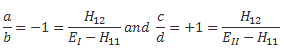My posts on the state transitions of an ammonia molecule weren’t easy, were they? So let’s try another two-state system. The illustration below shows an ionized hydrogen molecule in two possible states which, as usual, we’ll denote as |1〉 and |2〉. An ionized hydrogen molecule is an H2 molecule which lost an electron, so it’s two protons with one electron only, so we denote it as H2+. The difference between the two states is obvious: the electron is either with the first proton or with the second.

It’s an example taken from Feynman’s Lecture on two-state systems. The illustration itself raises a lot of questions, of course. The most obvious question is: how do we know which proton is which? We’re talking identical particles, right? Right. We should think of the proton spins! However, protons are fermions and, hence, they can’t be in the same state, so they must have opposite spins. Of course, now you’ll say: they’re not in the same state because they’re at different locations. Well… Now you’ve answered your own question. 🙂 However you want to look at this, the point is: we can distinguish both protons. Having said that, the reflections above raise other questions: what reference frame are we using? The answer is: it’s the reference frame of the system. We can mirror or rotate this image however we want – as I am doing below – but state |1〉 is state |1〉, and state |2〉 is state |2〉.

The other obvious question is more difficult. If you’ve read anything at all about quantum mechanics, you’ll ask: what about the in-between states? The electron is actually being shared by the two protons, isn’t it? That’s what chemical bonds are all about, no? Molecular orbitals rather than atomic orbitals, right? Right. That’s actually what this post is all about. We know that, in quantum mechanics, the actual state – or what we think is the actual state – is always expressed as some linear combination of so-called base states. We wrote:
|ψ〉 = |1〉C1 + |2〉C2 = |1〉〈1|ψ〉 + |2 〉〈2|ψ 〉
In terms of representing what’s actually going on, we only have these probability functions: they say that, if we would take a measurement, the probability of finding the electron near the first or the second proton varies as shown below:

If the |1〉 and |2〉 states were actually representing two dual physical realities, the actual state of our H2+ molecule would be represented by some square or some pulse wave, as illustrated below. [We should be calling it a square function but that term has been reserved for a function like y = x2.]

Of course, the symmetry of the situation implies that the average pulse duration τ would be one-half of the (average) period T, so we’d be talking a square wavefunction indeed. The two wavefunctions both qualify as probability density functions: the system is always in one state or the other, and the probabilities add up to one. But you’ll agree we prefer the smooth squared sine and cosine functions. To be precise, these smooth functions are:
- P1(t) = |C1(t)|2 = cos2[(A/ħ)·t]
- P2(t) = |C2(t)|2 = sin2[(A/ħ)·t]
So now we only need to explain A here (you know ħ already). But… Well… Why would we actually prefer those smooth functions? An irregular pulse function would seem to be doing a better job when it comes to modeling reality, doesn’t it? The electron should be either here, or there. Isn’t it?
Well… No. At least that’s why I am slowly starting to understand. These pure base states |1〉 and |2〉 are real and not real at the same time. They’re real, because it’s what we’ll get when we verify, or measure, the state, so our measurement will tell us that it’s here or there. There’s no in-between. [I still need to study weak measurement theory.] But then they are not real, because our molecule will never ever be in those two states, except for those ephemeral moments when (A/ħ)·t = n·π (n = 0, 1, 2,…). So we’re really modeling uncertainty here and, while I am still exploring what that actually means, you should think of the electron as being everywhere really, but with an unequal density in space—sort of. 🙂
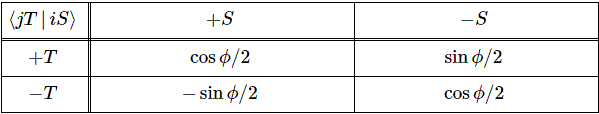
Now, we’ve learned we can describe the state of a system in terms of an alternative set of base states. We wrote: |ψ〉 = |I〉CI + |II〉CII = |I〉〈I|ψ〉 + |II〉〈II|ψ〉, with the CI, II and C1, 2 coefficients being related to each other in exactly the same way as the associated base states, i.e. through a transformation matrix, which we summarized as:

To be specific, the two sets of base states we’ve been working with so far were related as follows:

So we’d write: |ψ〉 = |I〉CI + |II〉CII = |I〉〈I|ψ〉 + |II〉〈II|ψ〉 = |1〉C1 + |2〉C2 = |1〉〈1|ψ〉 + |2 〉〈2|ψ 〉, and the CI, II and C1, 2 coefficients would be related in exactly the same way as the base states:

[In case you’d want to review how that works, see my post on the Hamiltonian and base states.] Now, we cautioned that it’s difficult to try to interpret such base transformations – often referred to as a change in the representation or a different projection – geometrically. Indeed, we acknowledged that (base) states were very much like (base) vectors – from a mathematical point of view, that is – but, at the same time, we said that they were ‘objects’, really: elements in some Hilbert space, which means you can do the operations we’re doing here, i.e. adding and multiplying. Something like |I〉CI doesn’t mean all that much: CI is a complex number – and so we can work with numbers, of course, because we can visualize them – but |I〉 is a ‘base state’, and so what’s the meaning of that, and what’s the meaning of the |I〉CI or CI|I〉 product? I could babble about that, but it’s no use: a base state is a base state. It’s some state of the system that makes sense to us. In fact, it may be some state that does not make sense to us—in terms of the physics of the situation, that is – but then there will always be some mathematical sense to it because of that transformation matrix, which establishes a one-to-one relationship between all sets of base states.
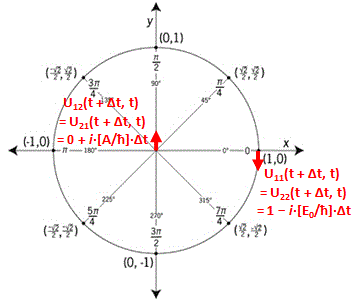
You’ll say: why don’t you try to give it some kind of geometrical or whatever meaning? OK. Let’s try. State |1〉 is obviously like minus state |2〉 in space, so let’s see what happens when we equate |1〉 to 1 on the real axis, and |2〉 to −1. Geometrically, that corresponds to the (1, 0) and (−1, 0) points on the unit circle. So let’s multiply those points with (1/√2, −1/√2) and (1/√2, 1/√2) respectively. What do we get? Well… What product should we take? The dot product, the cross product, or the ordinary complex-number product? The dot product gives us a number, so we don’t want that. [If we’re going to represent base states by vectors, we want all states to be vectors.] A cross product will give us a vector that’s orthogonal to both vectors, so it’s a vector in ‘outer space’, so to say. We don’t want that, I must assume, and so we’re left with the complex-number product, which projects our (1, 0) and (−1, 0) vectors into the (1/√2, −1/√2)·(1, 0) = (1/√2−i/√2)·(1+0·i) = √2−i/√2 = (1/√2, −i/√2) and (1/√2, 1/√2)·(−1, 0) = (1/√2+i/√2)·(−1+0·i) = −√2−i/√2 = (−1/√2, −i/√2) respectively.

What does this say? Nothing. Stuff like this only causes confusion. We had two base states that were ‘180 degrees’ apart, and now our new base states are only ’90 degrees’ apart. If we’d ‘transform’ the two new base states once more, they collapse into each other: (1/√2, −1/√2)·(1/√2, −1/√2) = (1/√2−i/√2)2 = −i = (0, −1) = (1/√2, 1/√2)·(−1/√2, −1/√2) = −i. This is nonsense, of course. It’s got nothing to do with the angle we picked for our original set of base states: we could have separated our original set of base states by 90 degrees, or 45 degrees. It doesn’t matter. It’s the transformation itself: multiplying by (+1/√2, −1/√2) amounts to a clockwise rotation by 45 degrees, while multiplying by (+1/√2, +1/√2) amounts to the same, but counter-clockwise. So… Well… We should not try to think of our base vectors in any geometric way, because it just doesn’t make any sense. So Let’s not waste time on this: the ‘base states’ are a bit of a mystery, in the sense that they just are what they are: we can’t ‘reduce’ them any further, and trying to interpret them geometrically leads to contradictions, as evidenced by what I tried to do above. Base states are ‘vectors’ in a so-called Hilbert space, and… Well… That’s not your standard vector space. [If you think you can make more sense of it, please do let me know!]
Onwards!
Let’s take our transformation again:
- |I〉 = (1/√2)|1〉 − (1/√2)|2〉 = (1/√2)[|1〉 − |2〉]
- |II〉 = (1/√2)|1〉 + (1/√2)|2〉 = (1/√2)[|1〉 + |2〉]
Again, trying to geometrically interpret what it means to add or subtract two base states is not what you should be trying to do. In a way, the two expressions above only make sense when combining them with a final state, so when writing:
- 〈ψ|I〉 = (1/√2)〈ψ|1〉 − (1/√2)〈ψ|2〉 = (1/√2)[〈ψ|1〉 − 〈ψ|2〉]
- 〈ψ|II〉 = (1/√2)〈ψ|1〉 + (1/√2)〈ψ|2〉 = (1/√2)[〈ψ|1〉 + 〈ψ|2〉]
Taking the complex conjugate of this gives us the amplitudes of the system to be in state I or state II:
- 〈I|ψ〉 = 〈ψ|I〉* = (1/√2)[〈ψ|1〉* − 〈ψ|2〉*] = (1/√2)[〈1|ψ〉 − 〈2|ψ〉]
- 〈II|ψ〉 = 〈ψ|II〉* = (1/√2)[〈ψ|1〉* + 〈ψ|2〉*] = (1/√2)[〈1|ψ〉 + 〈2|ψ〉]
That still doesn’t tell us much, because we’d need to know the 〈1|ψ〉 and 〈2|ψ〉 functions, i.e. the amplitudes of the system to be in state 1 and state 2 respectively. What we do know, however, is that the 〈1|ψ〉 and 〈2|ψ〉 functions will have some rather special amplitudes. We wrote:
- CI = 〈 I | ψ 〉 = e−(i/ħ)·EI·t
- CII = 〈 II | ψ 〉 = e−(i/ħ)·EII·t
These are amplitudes of so-called stationary states: the associated probabilities – i.e. the absolute square of these functions – do not vary in time: |e−(i/ħ)·EI·t|2 = |e−(i/ħ)·EII·t|2 = 1. For our ionized hydrogen molecule, it means that, if it would happen to be in state I, it will stay in state I, and the same goes for state II. We write:
〈 I | I 〉 = 〈 II | II 〉 = 1 and 〈 I | II 〉 = 〈 II | I 〉 = 0
That’s actually just the so-called ‘orthogonality’ condition for base states, which we wrote as 〈i|j〉 = 〈j|i〉 = δij, but, in light of the fact that we can’t interpret them geometrically, we shouldn’t be calling it like that. The point is: we had those differential equations describing a system like this. If the amplitude to go from state 1 to state 2 was equal to some real- or complex-valued constant A, then we could write those equations either in terms of C1 and C2, or in terms of CI and CII:

So the two sets of equations are equivalent. However, what we want to do here is look at it in terms of CI and CII. Let’s first analyze those two energy levels EI = E0 + A and EII = E0 − A. Feynman graphs them as follows:

Let me explain. In the first graph, we have EI = E0 + A and EII = E0 − A, and they are depicted as being symmetric, with A depending on the distance between the two protons. As for E0, that’s the energy of a hydrogen atom, i.e. a proton with a bound electron, and a separate proton. So it’s the energy of a system consisting of a hydrogen atom and a proton, which is obviously not the same as that of an ionized hydrogen molecule. The concept of a molecule assumes the protons are closely together. We assume E0 = 0 if the interproton distance is relatively large but, of course, as the protons come closer, we shouldn’t forget the repulsive electrostatic force between the two protons, which is represented by the dashed line in the first graph. Indeed, unlike the electron and the proton, the two protons will want to push apart, rather than pull together, so the potential energy of the system increases as the interproton distance decreases. So E0 is not constant either: it also depends on the interproton distance. But let’s forget about E0 for a while. Let’s look at the two curves for A now.
A is not varying in time, but its value does depend on the distance between the two protons. We’ll use this in a moment to calculate the approximate size of the hydrogen nucleus in a calculation that closely resembles Feynman’s calculation of the size of a hydrogen atom. That A should be some function of the interproton distance makes sense: the transition probability, and therefore A, will exponentially decrease with distance. There are a few things to reflect on here:
1. In the mentioned calculation of the size of a hydrogen atom, which is based on the Uncertainty Principle, Feynman shows that the energy of the system decreases when an electron is bound to the proton. The reasoning is that, if the potential energy of the electron is zero when it is not bound, then its potential energy will be negative when bound. Think of it: the electron and the proton attract each other, so it requires force to separate them, and force over a distance is energy. From our course in electromagnetics, we know that the potential energy, when bound, should be equal to −e2/a0, with e2 the squared charge of the electron divided by 4πε0, and a0 the so-called Bohr radius of the atom. Of course, the electron also has kinetic energy. It can’t just sit on top of the proton because that would violate the Uncertainty Principle: we’d know where it was. Combining the two, Feynman calculates both a0 as well as the so-called Rydberg energy, i.e. the total energy of the bound electron, which is equal to −13.6 eV. So, yes, the bound state has less energy, so the electron will want to be bound, i.e. it will want to be close to one of the two protons.
2. Now, while that’s not what’s depicted above, it’s clear the magnitude of A will be related to that Rydberg energy which − please note − is quite high. Just compare it with the A for the ammonia molecule, which we calculated in our post on the maser: we found an A of about 0.5×10−4 eV there, so that’s like 270,000 times less! Nevertheless, the possibility is there, and what happens when the electron flips over amounts to tunneling: it penetrates and crosses a potential barrier. We did a post on that, and so you may want to look at how that works. One of the weird things we had to consider when a particle crosses such potential barrier, is that the momentum factor p in its wavefunction was some pure imaginary number, which we wrote as p = i·p’. We then re-wrote that wavefunction as a·e−iθ = a·e−i[(E/ħ)∙t − (i·p’/ħ)x] = a·e−i(E/ħ)∙t·ei2·p’·x/ħ = a·e−i(E/ħ)∙t·e−p’·x/ħ. Now, it’s easy to see that the e−p’·x/ħ factor in this formula is a real-valued exponential function, with the same shape as the general e−x function, which I depict below.

This e−p’·x/ħ basically ‘kills’ our wavefunction as we move in the positive x-direction, across the potential barrier, which is what is illustrated below: if the distance is too large, then the amplitude for tunneling goes to zero.

So that’s what depicted in those graphs of EI = E0 + A and EII = E0 − A: A goes to zero when the interproton distance becomes too large. We also recognize the exponential shape for A in those graphs, which can also be derived from the same tunneling story.
Now we can calculate E0 + A and E0 − A taking into account that both terms vary with the interproton distance as explained, and so that gives us the final curves on the right-hand side, which tell us that the equilibrium configuration of the ionized hydrogen molecule is state II, i.e. the lowest energy state, and the interproton distance there is approximately one Ångstrom, i.e. 1×10−10 m. [You can compare this with the Bohr radius, which we calculated as a0 = 0.528×10−10 m, so that all makes sense.] Also note the energy scale: ΔE is the excess energy over a proton plus a hydrogen atom, so that’s the energy when the two protons are far apart. Because it’s the excess energy, we have a zero point. That zero point is, obviously, the energy of a hydrogen atom and a proton. [Read this carefully, and please refer back to what I wrote above. The energy of a system consisting of a hydrogen atom and a proton is not the same as that of an ionized hydrogen molecule: the concept of a molecule assumes the protons are closely together.] We then re-scale by dividing by the Rydberg energy EH = 13.6 eV. So ΔE/EH ≈ −0.2 ⇔ ΔE ≈ −0.2×13.6 = –2.72 eV. That basically says that the energy of our ionized hydrogen molecule is 2.72 eV lower than the energy of a hydrogen atom and a proton.
Why is it lower? We need to think about our model of the hydrogen atom once more: the energy of the electron was minimized by striking a balance between (1) being close to the proton and, therefore, having a low potential energy (or a low coulomb energy, as Feynman calls it) and (2) being further away from the proton and, therefore, lowering its kinetic energy according to the Uncertainty Principle ΔxΔp ≥ ħ/2, which Feynman boldly re-wrote as p = ħ/a0. Now, a molecular orbital, i.e. the electron being around two protons, results in “more space where the electron can have a low potential energy”, as Feynman puts it, so “the electron can spread out—lowering its kinetic energy—without increasing its potential energy.”
The whole discussion here actually amounts to an explanation for the mechanism by which an electron shared by two protons provides, in effect, an attractive force between the two protons. So we’ve got a single electron actually holding two protons together, which chemists refer to as a “one-electron bond.”
So… Well… That explains why the energy EII = E0 − A is what it is, so that’s smaller than E0 indeed, with the difference equal to the value A for an interproton distance of 1 Å. But how should we interpret EI = E0 + A? What is that higher energy level? What does it mean?
That’s a rather tricky question. There’s no easy interpretation here, like we had for our ammonia molecule: the higher energy level had an obvious physical meaning in an electromagnetic field, as it was related to the electric dipole moment of the molecule. That’s not the case here: we have no magnetic or electric dipole moment here. So, once again, what’s the physical meaning of EI = E0 + A? Let me quote Feynman’s enigmatic answer here:
“Notice that this state is the difference of the states |1⟩ and |2⟩. Because of the symmetry of |1⟩ and |2⟩, the difference must have zero amplitude to find the electron half-way between the two protons. This means that the electron is somewhat more confined, which leads to a larger energy.”
What does he mean with that? It seems he’s actually trying to do what I said we shouldn’t try to do, and that is to interpret what adding versus subtracting states actually means. But let’s give it a fair look. We said that the |I〉 = (1/√2)[|1〉 − |2〉] expression didn’t mean much: we should add a final state and write: 〈ψ|I〉 = (1/√2)[〈ψ|1〉 − 〈ψ|2〉], which is equivalent to 〈I|ψ〉 = (1/√2)[〈1|ψ〉 − 〈2|ψ〉]. That still doesn’t tell us anything: we’re still adding amplitudes, and so we should allow for interference, and saying that |1⟩ and |2⟩ are symmetric simply means that 〈1|ψ〉 − 〈2|ψ〉 = 〈2|ψ〉 − 〈1|ψ〉 ⇔ 2·〈1|ψ〉 = 2·〈2|ψ〉 ⇔ 〈1|ψ〉 = 〈2|ψ〉. Wait a moment! That’s an interesting reflection. Following the same reasoning for |II〉 = (1/√2)[|1〉 + |2〉], we get 〈1|ψ〉 + 〈2|ψ〉 = 〈2|ψ〉 + 〈1|ψ〉 ⇔ … Huh? No, that’s trivial: 0 = 0.
Hmm… What to say? I must admit I don’t quite ‘get’ Feynman here: state I, with energy EI = E0 + A, seems to be both meaningless as well as impossible. The only energy levels that would seem to make sense here are the energy of a hydrogen atom and a proton and the (lower) energy of an ionized hydrogen molecule, which you get when you bring a hydrogen atom and a proton together. 🙂
But let’s move to the next thing: we’ve added only one electron to the two protons, and that was it, and so we had an ionized hydrogen molecule, i.e. an H2+ molecule. Why don’t we do a full-blown H2 molecule now? Two protons. Two electrons. It’s easy to do. The set of base states is quite predictable, and illustrated below: electron a can be either one of the two protons, and the same goes for electron b.
We can then go through the same as for the ion: the molecule’s stability is shown in the graph below, which is very similar to the graph of the energy levels of the ionized hydrogen molecule, i.e. the H2+ molecule. The shape is the same, but the values are different: the equilibrium state is at an interproton distance of 0.74 Å, and the energy of the equilibrium state is like 5 eV (ΔE/EH ≈ −0.375) lower than the energy of two separate hydrogen atoms.
 The explanation for the lower energy is the same: state II is associated with some kind of molecular orbital for both electrons, resulting in “more space where the electron can have a low potential energy”, as Feynman puts it, so “the electron can spread out—lowering its kinetic energy—without increasing its potential energy.”
The explanation for the lower energy is the same: state II is associated with some kind of molecular orbital for both electrons, resulting in “more space where the electron can have a low potential energy”, as Feynman puts it, so “the electron can spread out—lowering its kinetic energy—without increasing its potential energy.”
However, there’s one extra thing here: the two electrons must have opposite spins. That’s the only way to actually distinguish the two electrons. But there is more to it: if the two electrons would not have opposite spin, we’d violate Fermi’s rule: when identical fermions are involved, and we’re adding amplitudes, then we should do so with a negative sign for the exchanged case. So our transformation would be problematic:
〈II|ψ〉 = (1/√2)[〈1|ψ〉 + 〈2|ψ〉] = (1/√2)[〈2|ψ〉 + 〈1|ψ〉]
When we switch the electrons, we should get a minus sign. The weird thing is: we do get that minus sign for state I:
〈I|ψ〉 = (1/√2)[〈1|ψ〉 − 〈2|ψ〉] = −(1/√2)[〈2|ψ〉 − 〈1|ψ〉]
So… Well… We’ve got a bit of an answer there as to what that the ‘other’ (upper) energy level of EI = E0 + A actually means, in physical terms, that is. It models two hydrogens coming together with parallel electron spins. Applying Fermi’s rules – i.e. the exclusion principle, basically – we find that state II is, quite simply, not allowed for parallel electron spins: state I is, and it’s the only one. There’s something deep here, so let me quote the Master himself on it:
“We find that the lowest energy state—the only bound state—of the H2 molecule has the two electrons with spins opposite. The total spin angular momentum of the electrons is zero. On the other hand, two nearby hydrogen atoms with spins parallel—and so with a total angular momentum ℏ—must be in a higher (unbound) energy state; the atoms repel each other. There is an interesting correlation between the spins and the energies. It gives another illustration of something we mentioned before, which is that there appears to be an “interaction” energy between two spins because the case of parallel spins has a higher energy than the opposite case. In a certain sense you could say that the spins try to reach an antiparallel condition and, in doing so, have the potential to liberate energy—not because there is a large magnetic force, but because of the exclusion principle.”
You should read this a couple of times. It’s an important principle. We’ll discuss it again in the next posts, when we’ll be talking spin in much more detail once again. 🙂 The bottom line is: if the electrons are parallel, then they won’t ‘share’ any space at all and, hence, they are really much more confined in space, and the associated energy level is, therefore, much higher.
Post scriptum: I said we’d ‘calculate’ the equilibrium interproton distance. We didn’t do that. We just gave them through the graphs, which are based on the results of a ‘detailed quantum-mechanical calculation’—or that’s what Feynman claims, at least. I am not sure if they correspond to experimentally determined values, or what calculations are behind, exactly. Feynman notes that “this approximate treatment of the H2+ molecule as a two-state system breaks down pretty badly once the protons get as close together as they are at the minimum in the curve and, therefore, it will not give a good value for the actual binding energy. For small separations, the energies of the two “states” we imagined are not really equal to E0, and a more refined quantum mechanical treatment is needed.”
So… Well… That says it all, I guess.
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/
Some content on this page was disabled on June 16, 2020 as a result of a DMCA takedown notice from The California Institute of Technology. You can learn more about the DMCA here:
https://wordpress.com/support/copyright-and-the-dmca/